See the Table of Materials for details related to all the materials and instruments used in this protocol. See Table 1 for a list of the solutions, buffers, and media used.
1. Recombinant yeast strain construction
- To construct a recombination-competent yeast strain, transform SbfI-digested plasmid K238 into a yeast strain with integrated LexA-binding sites and RS recombination sites at the locus of interest13,14.
NOTE: The transformed SbfI restriction fragment contains the required expression cassette for constitutive expression of the LexA-TAP fusion protein and R-recombinase together with homologous sequences of yeast chromosome I for genomic integration of the expression cassette by homologous recombination (Figure 1). - To construct a control strain, take an isogenic yeast strain lacking integrated RS and LexA-binding sites, and transform it with SbfI-digested K238 plasmid under the same conditions as used for the recombination-competent strain.
- Select the competent yeast cells based on the LEU2 selection marker on SCD-LEU agar plates14.
2. Coupling IgG antibodies to epoxy-activated magnetic beads
NOTE: Couple IgG antibodies to epoxy-activated magnetic beads according to the following published protocol11.
- Suspend 300 mg of epoxy-activated beads in 10 mL of 50% acetone (300 mg corresponds to ~5.1 × 1010 beads) in a 50 mL conical tube. Shake vigorously on a vortex mixer.
- Centrifuge the tube containing the beads at 820 × g and 4 °C for 2 min. Remove the supernatant.
- Wash the beads 3x with 20 mL of 0.1 M sodium phosphate buffer (pH 7.4). Remove the supernatant after each washing step by centrifugation as described in step 2.2.
- Suspend the beads in 16 mL of 0.1 M sodium phosphate buffer (pH 7.4), and rotate gently in a hybridization oven for 5 min at room temperature.
- Dissolve the rabbit IgGs (100 mg) in 7 mL of dH2O (final concentration: 14 mg/mL).
- Centrifuge the IgG suspension at 13,000 × g and 4 °C for 10 min to clarify the suspension.
- Transfer 3.5 mL of the supernatant (corresponding to 50 mg of rabbit IgGs) to a new 50 mL conical tube.
- Dilute the IgG solution with 9.85 mL of 0.1 M sodium phosphate buffer (pH 7.4), followed by the dropwise addition of 6.65 mL of 3 M ammonium sulfate in 0.1 M sodium phosphate (pH 7.4) under gentle mixing.
NOTE: Avoid adding ammonium sulfate quickly to the solution as a high local concentration of the salt will cause the precipitation of the rabbit IgGs. - Centrifuge the IgG solution at 820 × g and 4 °C for 3 min, and add the resulting supernatant to the magnetic bead suspension.
- Incubate the tube overnight or at least for 18 h at 30 °C with gentle rotation in a hybridization oven.
- Remove the supernatant as described in step 2.2.
- Wash the beads with 20 mL of 100 mM glycine-HCl (pH 2.5). Remove the solution rapidly as described in step 2.2 to avoid the denaturation of the IgG polypeptides.
- Wash the beads once with 20 mL of 10 mM Tris-HCl (pH 8.8). Aspirate the supernatant as described in step 2.2.
- Add 20 mL of 0.1M triethylamine solution for 5-10 min under gentle rotation to inactivate the residual reactive epoxy groups. Remove the supernatant as described in step 2.2.
- Wash the beads 4x with 20 mL of PBS (pH 7.4) for 5 min with gentle rotation. Remove the supernatant as described in step 2.2 after each washing step.
- Wash the beads 2x with 20 mL of PBS (pH 7.4) with 0.5% Triton X-100 (w/v) for 5 min and 15 min each under gentle rotation in a hybridization oven. Remove the supernatant as described in step 2.2.
- Suspend the beads in a final volume of 16 mL of PBS (pH 7.4) with 0.02% sodium azide (w/v). Store as 1 mL aliquots at 4 °C until use.
3. Yeast cell culture and harvesting
- Inoculate control and recombination competent yeast strains from YPD plates in 5 mL of YPR medium, and incubate overnight at 30 °C and 200 rpm.
- Inoculate 2 mL of this culture into 100 mL of YPR medium, and incubate overnight at 30 °C and 200 rpm.
- For each strain, dispense 1,800 mL of autoclaved YP medium and 200 mL of autoclaved raffinose solution (20% w/v) into each of two 5 L Erlenmeyer flasks (4 L of culture medium in total for each strain, divided into two flasks).
- Inoculate the growing yeast cells at OD600 0.2 in their respective medium, and incubate as described in step 3.1 for approximately 6 h or until the cells reach the desired OD600 of 1.0. To ensure normal growth of the cells, free from any contamination, check the OD at 2 h intervals.
- Add 200 mL of galactose (20% w/v) to induce site-specific recombination.
- Add 110 µL (50 ng/mL) of YMFA (at the same time as the galactose, step 3.5) to arrest the cells in the G1 phase, and incubate for 2 h.
NOTE: The addition of YMFA to the cells depends on the experimental conditions and biological question to be addressed. Precisely, for this experiment, the cells are arrested in G1 phase to obtain a homogenous cell population with licensed replication origins; in this process, the pre-replicative complex binds to the origins in the G1 phase prior to the initiation of DNA replication in the S-phase. - Transfer the cell suspension into 1 L centrifuge buckets, and centrifuge at 6,000 × g and 4 °C for 10 min. Discard the supernatant, and resuspend the cell pellets in a total volume of 10-15 mL of dH2O.
- Seal a 25 mL syringe with a Luer plug, and place it inside a 50 mL conical tube filled with water. Transfer the cell suspension to the syringe assembly, and centrifuge at 2,397 × g for 10 min at room temperature. Discard the supernatant.
- Remove the Luer plug from the syringe, and extrude the cells into liquid nitrogen to form cell "spaghetti".
NOTE: Using 4 L of culture medium provides a wet weight of final cell pellets of 7-10 g. - Transfer the frozen cell spaghetti to a 50 mL conical tube, and store at −80 °C until further use.
4. Chromatin locus purification
NOTE: See Figure 2 for a schematic overview of the steps involved in this locus-specific chromatin purification protocol.
- Cool down a commercial coffee grinder by grinding 30-50 g of dry ice by vigorously shaking the coffee grinder 2x for 30 s each time. Once cooled, discard the dry ice powder.
- Mix 3 g of frozen yeast cell spaghetti with ~30-50 g of dry ice in the precooled coffee grinder.
- Seal the junction between the cap and the grinder with parafilm to prevent the loss of yeast cell powder during milling.
- Mill the yeast cell-dry ice mix 10x for 30 s each time, with 30 s intervals between each round (total time estimate: ~10 min). To avoid the dry ice and cell powder sticking to the walls of the coffee grinder, tap the sides of the coffee grinder continuously during milling.
- Transfer the resulting yeast cell-dry ice powder to a plastic beaker using a clean and dry spatula.
- Keep the beakers at room temperature to evaporate the dry ice.
- Once the dry ice evaporates, add 2.25 mL of MB buffer with 1x protease and phosphatase inhibitors (750 µL/g of yeast cells) to the yeast cell powder.
- Pipette the cell-buffer mixture vigorously to ensure the complete suspension of the cells with the buffer, and transfer to a 15 mL conical tube.
- Take samples for DNA (0.1%) and protein (0.05%) analysis from the resulting cell suspension (crude cell extracts [CCE]). Store at −20 °C until further processing (described in section 7).
- Transfer the cell suspension into low-binding 2 mL reaction tubes, and centrifuge at 21,130 × g for 30 min at 4 °C to separate the cell debris from the crude cell lysate.
- Pool the supernatants from all the tubes into one 15 mL conical tube.
- Take samples from the supernatant (input [IN]) for DNA (0.1%) and protein analysis (0.05%). Store at −20 °C until further processing (described in section 7).
- Equilibrate 500 µL of IgG-coupled magnetic bead slurry (for each yeast strain) by washing 2x with 500 µL of cold MB buffer with 1x protease and phosphatase inhibitors for 5 min each at 4 °C on rotation at 20 rpm. Discard the supernatant from the beads using a magnetic rack after each washing step.
- Incubate the IgG-coupled magnetic beads in 500 µL of cold MB buffer with 1x protease and phosphatase inhibitors for 1 h at 4 °C on rotation at 20 rpm. Discard the supernatant from the beads using a magnetic rack.
- Mix the equilibrated magnetic bead slurry with the cell lysate, and incubate with rotation at 20 rpm for 2 h at 4 °C.
- Transfer the complete magnetic bead-cell lysate suspension to low-binding reaction tubes.
- Separate the magnetic beads, now carrying the chromatin rings of interest, from the cell lysate using a magnetic rack.
- Transfer the remaining flow-through (FT) from each tube to a fresh 15 mL conical tube, and take samples for DNA (0.1%) and protein (0.05%) analysis. Store at −20 °C until further processing (described in section 7).
- Suspend the magnetic beads from each tube in 300 µL of cold MB buffer, and pool the beads into one reaction tube. Wash 5x for 10 min each with 750 µL of cold MB buffer with 1x protease and phosphatase inhibitors at 4 °C on rotation at 20 rpm. Perform the final wash with 750 µL of cold MB buffer without protease and phosphatase inhibitors.
- Suspend the beads in 40 µL of cold MB buffer without protease and phosphatase inhibitors.
NOTE: The final volume of eluate can be adjusted according to its anticipated downstream application. The TEV elution usually works efficiently within the range of 100-300 µL.
5. TEV protease-mediated elution
- To release LexA-CBP chromatin ring complexes from the beads, incubate the beads with 2 µL of 6x His-tagged recombinant TEV protease overnight at 4 °C and 450 rpm in a thermomixer.
- Separate the beads from the final eluate using a magnetic rack. Transfer the eluate, containing cleaved chromatin rings, to a new 1.5 mL reaction tube.
- Keep the tubes on a magnetic rack again to separate any residual beads from the final eluate.
- Resuspend the beads (TB) in 750 µL of cold AC buffer, and take samples for DNA (0.1%) and protein (0.05%) analysis. Store at −20 °C until further processing (described in section 7).
- From the final eluate (TE), take samples for DNA (0.5%) and protein (0.25%) analysis. Store at −20 °C until further processing (described in section 7).
NOTE: Due to the low volume of the final eluate, the percentage of the samples collected for DNA and protein analysis is increased to obtain a better representation of their protein and DNA content during analysis.
6. Denaturation elution
- Wash the beads 2x in 750 µL of cold AC buffer for 20 min each time at 4 °C on rotation at 20 rpm.
- To extract the remaining bound LexA-chromatin complexes, carry out denaturation elution by adding 500 µL of 0.5M NH4OH to the beads, and incubate for 30 min at room temperature.
- Separate the beads from the suspension using a magnetic rack, and transfer the eluate to a low-binding reaction tube.
- Incubate the beads again as in step 6.2 to recover the maximum chromatin loci in the eluate.
- Separate the beads from the suspension using a magnetic rack, and pool the resulting eluate into the same tube containing the eluate from step 6.3.
- Resuspend the beads (DB) in 750 µL of dH2O, and take samples for DNA (0.1%) and protein (0.05%) analysis.
- From the final eluate (DE), take samples for DNA (0.5%) and protein (0.25%) analysis.
7. DNA and protein analysis
- DNA analysis
- Sample preparation
- To the DNA samples, add 100 µL of IRN buffer, and increase the volume with dH2O to a final volume of 200 µL.
- Add 1 ng of K071 spike-in plasmid DNA.
- Add 1 µL of RNAse A (10 mg/mL), and incubate at 37 °C for 1 h.
- Add 5 µL of proteinase K (10 mg/mL) and 10 µL of SDS (20%), and incubate for 1 h at 56 °C.
- Add 200 µL of phenol/chloroform/isopropyl alcohol (25:24:1), and vortex thoroughly 2x for 10 s each time.
- Centrifuge the samples at 21,130 × g for 7 min to separate the organic and aqueous phases.
- Transfer the supernatant to new 1.5 mL tubes containing 1.5 µL of glycogen (5 mg/mL) and 2.5x 100% ethanol.
- Incubate the tubes at −20 °C for a minimum of 2 h and a maximum of overnight to precipitate the DNA.
- Centrifuge at 21,130 × g, 4 °C for 45 min. Discard the supernatant.
- Add 150 µL of 70% ethanol without resuspending the DNA pellet, and centrifuge again at 21,130 × g at 4 °C for 10 min.
- Dry the DNA pellets at room temperature for 10-15 min, and resuspend in 40 µL of dH2O.
- Perform restriction enzyme digestion to linearize the DNA isolated in step 7.1.1.11 with HpaI restriction endonuclease. For a 50 µL reaction mix, incubate 40 µL of isolated DNA + 2 µL of HpaI enzyme + 5 µL of restriction enzyme buffer + 3 µL of dH2O overnight at 37 °C.
- qPCR analysis
- Dilute the restriction-digested DNA at a 1:5 ratio (10 µL of the restriction-digested DNA obtained from step 7.1.1.12 in 40 µL of dH2O).
- Use primer pairs designed for the ARS316 region, PDC1, and spike-in plasmid DNA.
- Run qPCR using the program given in Table 2.
- Analyze the qPCR results by relative quantification using the ARS316 region and spike-in plasmid DNA as targets and PDC1 (or any other gene or genomic region) as the reference locus.
- Assess the ARS316 locus recovery percentage over PDC1 by normalizing the ARS316 and PDC1 primer relative quantification values by the percentage of the fraction volume taken for each sample. For instance, multiply 0.1% for the flow-through by a factor of 1,000 and 0.5% for the TEV eluate by a factor of 200.
- Normalize the ARS316 and PDC1 recovery percentages with the spike-in plasmid DNA relative quantification values.
- Assess the enrichment of the ARS316 locus over the total chromatin by evaluating the obtained relative quantification target/reference values using equation (1).
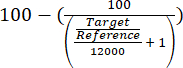 (1)
(1)
- Southern blot analysis
- To 20 µL of restriction enzyme-digested DNA sample, add 5 µL of gel loading dye.
- Run the samples in a 1% agarose gel at 110 V for ~3 h.
- Transfer the gel to a tray, and soak it in depurination solution with gentle shaking for 20 min.
- Rinse the gel with dH2O, and soak it in denaturation solution for 15 min with shaking. Discard the denaturation solution.
- Soak the gel in denaturation solution for 15 min with gentle shaking.
- Rinse the gel with dH2O, and wash it 2x for 15 min each time with the transfer buffer with gentle shaking.
- Fill up a tray with 1-1.5 L of transfer buffer, and place it on a stable platform.
- Cut a long piece of Whatman paper, and place it on the platform in such a way that the two ends of the paper are soaked in the transfer buffer.
- Cut one strip of positive nylon membrane and four strips of Whatman paper equal to the size of the gel.
- Place two pieces of Whatman paper soaked in the transfer buffer on the platform.
- Place the gel face down on top of the pieces of Whatman paper.
- On top of the gel, place the positive nylon membrane, followed by the remaining two pieces of Whatman paper soaked in transfer buffer. Make sure to keep the stack wet with the transfer buffer.
- Remove any trapped air bubbles by rolling them off using a glass rod.
- Place a stack of paper towels on top of the assembled stack, followed by a 0.5 kg object (e.g., a glass bottle).
- Leave the assembly for overnight transfer at room temperature.
- After transfer, UV-crosslink the membrane with a total energy output of 1,200 J/cm2.
- For the detection of the ARS316 locus, perform southern blot hybridization using radioactive probes synthesized using the referenced DNA labeling system.
NOTE: Perform all the steps hereafter on ice until mentioned otherwise. - In a low-binding reaction tube, dilute 25-40 ng of PCR fragment (probe) in 19 µL of dH2O, and incubate at 95 °C for 5 min. Cool immediately on ice.
- Add 1 µL each of 500 µM dCTP, 500 µM dGTP, and 500 µM dTTP to the tube.
- Add 20 µL of the buffer containing random nucleotide hexamers provided with the kit.
- Add 5 µL of the radioactively labeled nucleotide [α-32P] dATP to the tube. Perform all the steps involving radioactively labeled material under a radioactivity-approved protective environment.
- Add 1 µL of Klenow fragment, and incubate at 37 °C on a thermomixer for 15 min for single-stranded DNA synthesis.
- Add 5 µL of stop buffer to stop the reaction.
- Centrifuge a size exclusion column for 2 min at 1,500 × g to remove the storage buffer. Discard the flow through, and place the column in a fresh tube.
- Pass the probe mixture through the column to remove unincorporated radioactive nucleotides. Centrifuge for 30 s at 1,500 × g to collect the probe.
- Add 150 µL of salmon sperm DNA (1:100) to block the non-specific binding of the probe to the membrane.
- Incubate the probe mixture at 95 °C for 5 min to denature the double-stranded DNA.
- Snap-freeze on ice for a few seconds, and centrifuge for a few seconds at 500 × g to bring down the water droplets condensed on the lid.
- Briefly wash the southern blot membrane with the hybridization buffer for 5-10 min with rotation.
- Pre-hybridize the membrane with the hybridization buffer for 1 h at 55 °C with rotation. Discard the hybridization buffer.
- Add 15 mL of fresh hybridization buffer (pre-warmed at 55 °C) to the membrane along with the prepared probe. Incubate the membrane overnight at 55 °C with rotation.
- Perform post-hybridization washes 2x for 15 min each with 0.3x SSC, 0.1% SDS at 55 °C with rotation.
- Perform post-hybridization washes 2x for 15 min each with 0.1x SSC, 0.1% SDS at 55 °C with rotation.
- Perform post-hybridization washes 2x for 15 min each with 0.1x SSC, 1.5% SDS at 55 °C with rotation.
- Remove the membrane from the hybridization tube, and expose the membrane to a phosphorimager screen for up to 3 days to obtain the radioactive imprints on the film.
- Acquire images of the film using a phosphorimaging laser scanning system.
- Sample preparation
- Protein analysis
- Sample preparation
- Adjust all the protein samples to their final volumes (200 µL, 100 µL, 100 µL, 20 µL, and 20 µL for the crude cell extract, input, flow-through, beads, and eluate samples, respectively) by adding 1 µL of β-mercaptoethanol, PAGE LDS sample buffer (1x), and dH2O.
- Denature the samples at 95 °C for 5 min.
- Western blot analysis
NOTE: Perform SDS-PAGE and western blotting using standard protocols15,16.- Load 15 µL of each sample into each well of a 1.5 mm SDS-PAGE gel.
- Perform immunostaining of the blot using PAP (1:2,000) and LexA (1:1,000) antibodies with overnight incubation at 4 °C. Dilute in 5% dry milk/PBST solution.
NOTE: After PAP analysis, the blot can be stripped using a mild stripping protocol17 prior to immunostaining with the second LexA antibody.
- Sample preparation
The purification of the ~1.4 kb ARS316 chromatin domain was mediated by the constitutively expressed LexA-TAP adapter protein. To serve as a negative control, we conducted purifications using an isogenic strain that expresses LexA-TAP but does not contain integrated RS and LexA-binding sites. Figure 3 illustrates the DNA analysis outcome from a standard purification experiment performed on both the control and a recombination-competent strain targeting the ARS316 locus. After DNA isolation and restriction enzyme digestion to linearize the circular DNA molecules, qPCR analysis was performed to assess the purification efficiency of the ARS316 target locus over the unrelated genomic control locus PDC1.
Furthermore, the enrichment of the ARS316 locus over the total chromatin was quantified (Figure 3A,B). The negative control strain showed no recovery of the ARS316 or PDC1 loci in any of the fractions except for the crude cell extract, input, and flow-through, suggesting that no specific enrichment could be observed in the TEV and denaturing elutions. In contrast, the ARS316 locus was quantitatively depleted in the flow-through in the recombination strain and could be recovered on the beads after TEV elution and in the denaturing elution. After TEV elution, the beads showed a higher recovery percentage of the ARS316 locus as the TEV cleavage efficiency was not 100%. This resulted in a fraction of chromatin rings remaining bound to the beads. The TEV protease cleavage efficiency can be improved by increasing the elution volume above 100 µL. In contrast, the denaturing elution showed 80%-90% recovery of the ARS316 locus in the recombination strain (Figure 3A), corresponding to the high enrichment of ARS316 molecules when factoring in the size of the yeast genome (Figure 3B).
Southern blot was additionally performed to validate the qPCR results. Due to its high sensitivity, it can detect relatively lower concentrations of target DNA, thus allowing for quantitative analysis of the samples. The results obtained from the southern blot analysis using radioactively labeled probe confirmed the specific enrichment of the ARS316 locus in the fractions collected from the recombination strain but not in the control strain fractions (Figure 3C). In the recombination strain, higher enrichment of the ARS316 locus was observed based on the signal intensity in the TEV beads and the denaturation elution samples, concordant with the qPCR results. Similarly, the weak signals observed in the TEV elution and denaturation beads samples are concordant with the observed low enrichment of the ARS316 locus in these fractions (Figure 3C).
The LexA-TAP cleavage and pulldown efficiencies were also assessed by western blot analysis. The LexA-TAP protein has a molecular weight of ~80 kDa (Figure 4A). In both the recombination and control strains, western blot analysis was performed using the PAP antibody, which targets the protein A moiety of LexA-TAP in the purification samples. As anticipated, the results demonstrated near-total depletion of LexA-TAP in the flow-through, with recovery observed in the final bead and elution fractions (Figure 4B[i]). The PAP antibody also detects the small protein A fragment of ~15 kDa that remains on the beads after TEV elution. The same western blot was stripped and immunostained with an antibody against the LexA moiety of LexA-TAP (Figure 4B[ii]), showing complementary results (Figure 4B[ii]). The strong band at ~50 kDa in both blots indicates the IgG heavy chain coupled to the magnetic beads, which cross-hybridizes with the primary/secondary antibody conjugates (Figure 4B[ii]).
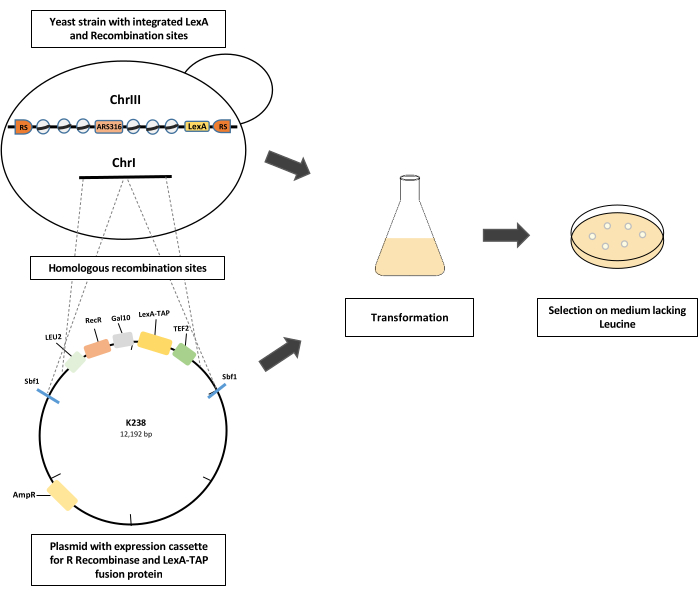
Figure 1: Schematics of yeast strain construction. A modified yeast strain with integrated LexA and recombination sites on chromosome III is transformed with a plasmid (K238) with expression cassettes for the constitutive expression of the LexA-TAP fusion protein using the TEF2 promoter and the galactose-inducible expression cassette for R recombinase (RecR) expression using a GAL10 promoter. The plasmid is linearized by SbfI restriction digestion, thereby creating homologous recombination arms for the integration of the expression cassette into an intergenic location on chromosome I. Competent cells are selected based on the LEU2 selection marker on the growth medium lacking leucine. Please click here to view a larger version of this figure.
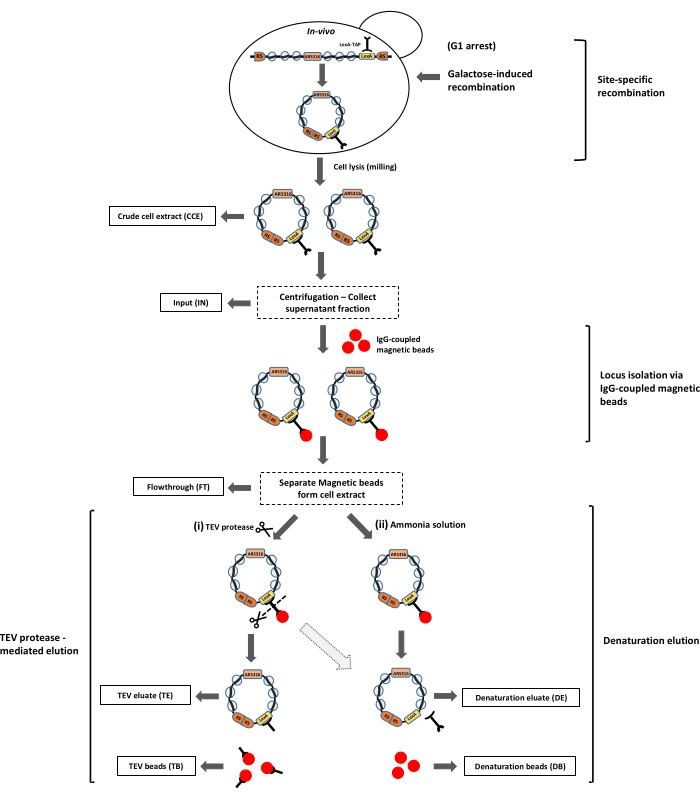
Figure 2: Purification of the ARS316 chromatin locus from the yeast Saccharomyces cerevisiae. The ARS316 locus is excised in the form of chromatin rings from its genomic location in yeast cells by galactose-induced site-specific recombination. These chromatin rings are isolated from the crude cell extract using the LexA-TAP affinity tag bound to IgG-coupled magnetic beads. Chromatin rings can be released from the beads by two methods: i) TEV protease-mediated cleavage, or ii) denaturation elution using ammonium solution. The dotted arrow represents the possibility of performing denaturation elution after TEV protease-mediated elution to release the remaining bead-bound chromatin rings, as the TEV protease cleavage efficiency is not 100%. The outlined boxes represent the samples obtained at different steps of the purification for protein and DNA analyses. Abbreviation: RS = recombination sites. Please click here to view a larger version of this figure.
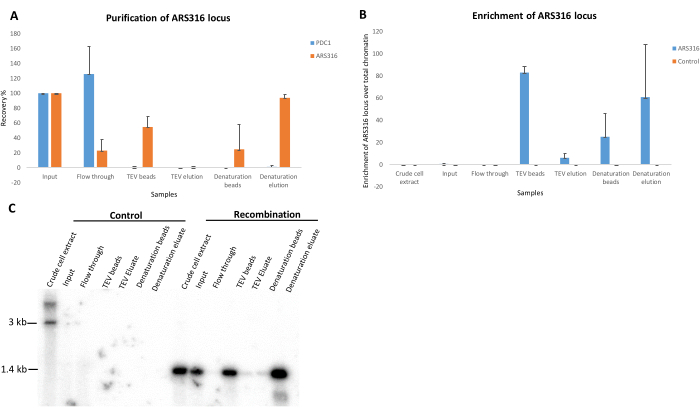
Figure 3: DNA analysis to evaluate the ARS316 locus purification. (A) Bar graph showing the purification of the ARS316 locus and an unrelated region, PDC1, in a series of DNA samples collected during ARS316 chromatin locus purification from the recombination yeast strain. (B) Bar graph showing the enrichment of the ARS316 locus over total chromatin in a series of DNA samples collected during ARS316 chromatin locus purification from control and recombination yeast strains. (C) Southern blot image showing the enrichment of the ARS316 locus in a series of DNA samples collected during ARS316 chromatin locus purification from control and recombination yeast strains. Please click here to view a larger version of this figure.
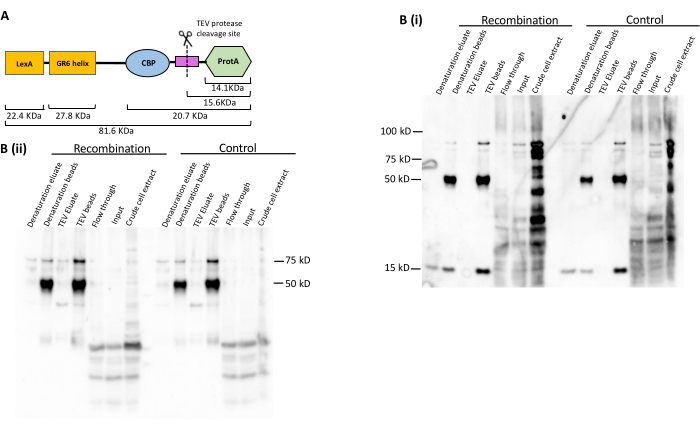
Figure 4: Protein analysis to evaluate LexA-TAP pull-down and cleavage efficiency. (A) Cartoon representing the different protein domains of the LexA-TAP fusion protein, along with the size of each domain. (B) Western blot images showing immunostaining against (i) PAP and (ii) LexA in a series of protein samples collected during ARS316 chromatin locus purification from control and recombination yeast strains. Please click here to view a larger version of this figure.
Figure 5: Flow diagram highlighting the potential downstream applications of chromatin domains purified using LexA-TAP affinity purification. Please click here to view a larger version of this figure.
Table 1: A list of the solutions and media used in this protocol. Please click here to download this Table.
Table 2: Quantitative PCR program. Please click here to download this Table.
