Following the cotransfection of HIViGFPΔEnv and psPAX2, all of the grids had minimal tearing in the carbon layer. Grids were imaged using phase light microscopy and fluorescent light microscopy 24 h after incubation with the transfection reagent (Figure 2). Cells on both the mock grids and the co-transfected grids contained viable cells in multiple grid squares.
psPAX2 codes for all structural and enzymatic proteins of HIV-1 without any fluorescence tagging. HIViGFPΔEnv is similar to psPAX2 but with codes for a GFP-tagged HIV Gag protein. Both plasmids are ΔEnvelope. The cotransfection results in native-like assembly and budding of fluorescent HIV-1 particles, making this a great system for CLEM studies of HIV in Biosafety Level 1 condition. The co-transfected grids showed a subset of cells exhibiting green fluorescence, indicating successful cotransfection. No cells on the mock grid exhibited fluorescence, further validating the cotransfection using HIViGFPΔEnv and psPAX2. After viewing the grids using light-based microscopy, grids were plunge frozen and moved to long-term storage in liquid nitrogen.
Figure 3 depicts results from grids produced using the same experimental method but utilizing slightly different plasmid constructs. U2OS-containing grids were co-transfected using different HIV clones (HIVmCherryΔEnv, and NL4-3ΔEnvGFP at a 1:6 ratio). Since a higher mass of fluorescently tagged plasmids were used, these grids enabled the observation of a larger number of transfected cells, providing an advantage while capturing images using cryoCLEM and cryoET. Using cryoCLEM, full grid atlases were generated for each grid using cryofluorescence microscopy to record the locations of all co-transfected cells. With the locations of cells known, cryoET was performed. A full low-magnification grid atlas was collected and overlaid with the fluorescent atlas collected at cryofluorescence (Figure 3A). Cryotomograms were collected at cellular sites capturing intricate details of the viral life cycle, including the assembly and budding of HIV from cells (Figure 3B).
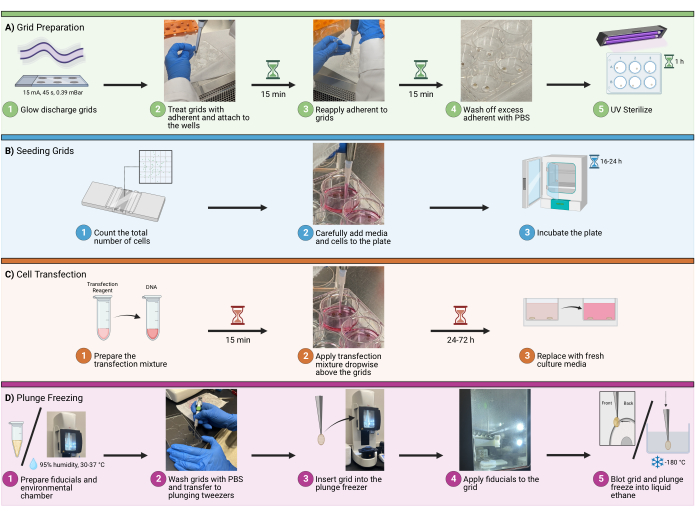
Figure 1: Cell seeding on grids workflow. A schematic depicting the overall procedure to seed cells on cryoEM grids. The process is divided into four major steps, including (A) preparing grids in the wells for seeding, (B) adding the appropriate amount of cells to each well, (C) the optional transfection of cells for fluorescent imaging, and (D) the plunge freezing of grids to allow for vitrification of the sample. Please click here to view a larger version of this figure.

Figure 2: Cotransfection of U2OS using HIViGFPΔEnv and psPAX2. U2OS cells were co-transfected with GFP-containing HIViGFPΔEnv and psPAX2 in a 1:3 ratio. Grids were imaged by phase contrast and fluorescence microscopy. Cells that are shown to have GFP expression indicate successful cotransfection. Scale bar: 500 µm. Scale bar (insets): 100 µm. Please click here to view a larger version of this figure.
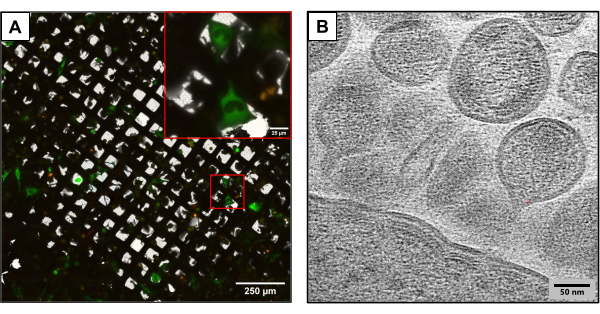
Figure 3: Potential downstream cryo-based methods. (A) A cryoCLEM image with co-transfected U2OS cells. Cells in green represent HIV-producing cells and are used to measure cotransfection success. Red puncta represent mCherry tagged HIV-1 Gag. Scale bar: 250 µm. Scale bar (inset): 25 µm (B) A cryoET image of multiple HIV particles budding from the plasma membrane of U2OS cells. Scale bar: 50 nm Please click here to view a larger version of this figure.
