Here, we utilize flow cytometry as a powerful tool to investigate the release of miRNA from the cells into EVs. Using this protocol, flow cytometry analysis of cells transfected with cell-miRNA and EV-miRNA revealed a sequential decrease of fluorescence signal corresponding to EV-miRNA, while the signal corresponding to the cell-miRNA was retained in the cells. To ensure that fluorophore conjugation does not interfere with miRNA release into EVs, miR-451a was conjugated to two separate fluorophores (i.e., Alexa fluor 488 and Alexa fluor 750), and cell retention was evaluated. The results indicate minimal variability between the detection of the separate fluorophore signals post-transfection, validating the reliability of this approach (Figure 3A,B).
Because various reasons could explain the loss of fluorescently tagged miR-451a from the cells, including RNA degradation, it was critical to verify that cellular loss coincided with EV abundance. Using flow cytometry, accumulation of miR-451a conjugated to Alexa fluor-488 in EVs was observed in a time-dependent manner, providing evidence of efficient packaging and eventual release of the miRNA into EVs. In contrast, the fluorescence signal corresponding to the cell retained miRNA, cel-miR-67 was not detected in the EVs (Figure 4). These findings using this new approach were validated using qRT-PCR, which revealed a significantly higher EV/cell expression ratio for miR-150, an additional EV-miRNA, when compared to the cell-miRNA, cel-miR-67 (Figure 5).
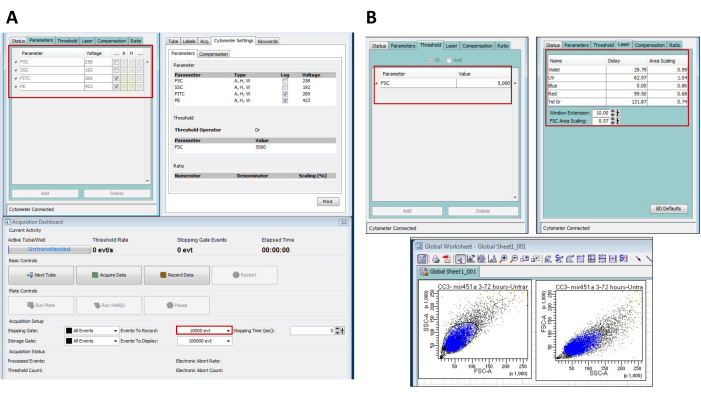
Figure 1: Set up of flow cytometer parameters for detection of signals in cells. (A) For the analysis of cell fluorescence, using the software, the highlighted voltages were set up for the parameters indicated. The instrument was set up to record 10,000 cells per sample. The Inspector View confirms the parameters set up for detection and analysis. (B) For the analysis of cell fluorescence, the Threshold for the FSC parameter was set at 5,000 under the cytometer settings tab of the software. The top right panel shows the laser settings for the data capture. The bottom panels show an active global worksheet during the progress of the detection of cells. The samples were run at a medium flow and captured while establishing FSC-A and SSC-A as x-axis and y-axis parameters, respectively. The gating was set using a drawing polygon tool around the population of interest. The gating might be different for another cell line of interest depending upon the diversity of the cell population. Please click here to view a larger version of this figure.
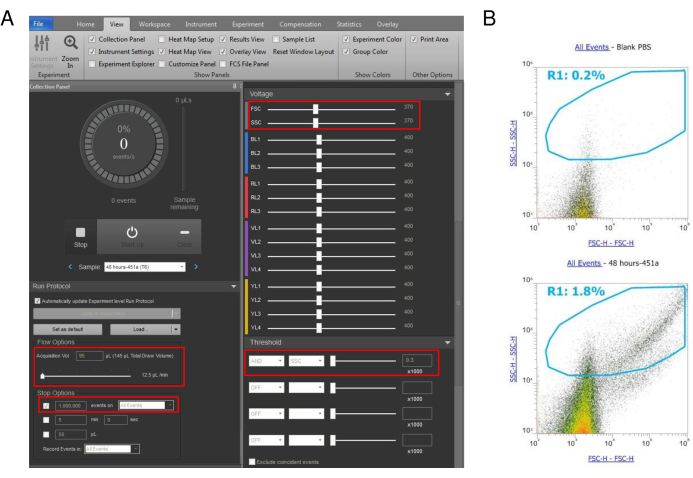
Figure 2: Set up of flow cytometry parameters for signal detection in EVs. (A) For the detection and downstream analysis of EVs, the acquisition volume was set at 95 µL with the flow speed set at 12.5 µL/min. The chosen threshold for EV detection was 0.3 x1000 under the instrument settings tab of the software based upon the calibration beads used. Voltages for FSC, SSC, BL1, and YL1 were 370, 370, 400 and 400, respectively. (B) Setup of gating for capturing heterogenous EV population and distinguishing EVs from the background noise of the instrument. A blank PBS sample (top) was used as a negative control to resolve background noise from the EV population. EV sample (bottom) shows that the population of interest is only 1.8% of the total population because the sample is heterogeneous with overlapping particles in the size range. However, stringent gating was chosen to ensure the detection of the EV population of interest. Please click here to view a larger version of this figure.
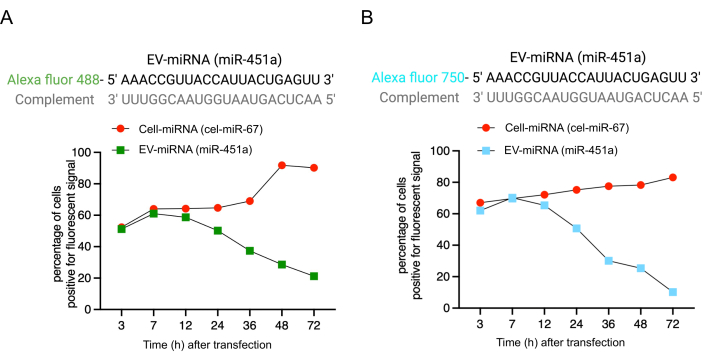
Figure 3: Signal detection in cells through flow cytometry. (A) Fluorescence corresponding to EV-miRNA and cell-miRNA was detected in cells at times indicated between 7 h and 72 h following transfection. The signal corresponding to EV-miR-451a (Alexa fluor-488) diminished with time in comparison to the signal from the cell-retained-miRNA, cel-miR-67. (B) Fluorescence corresponding to EV-miR-451a labeled with Alexa fluor-750 showed the same trend as the Alexa Fluro-488 labeled miR-451a, indicating that the fluorophore does not impact the retention of miRNAs by the cells. Overall, the representative results indicate that the transfected miRNA behaves similarly to the endogenous miRNA based on the export of the miRNA into EVs. Please click here to view a larger version of this figure.
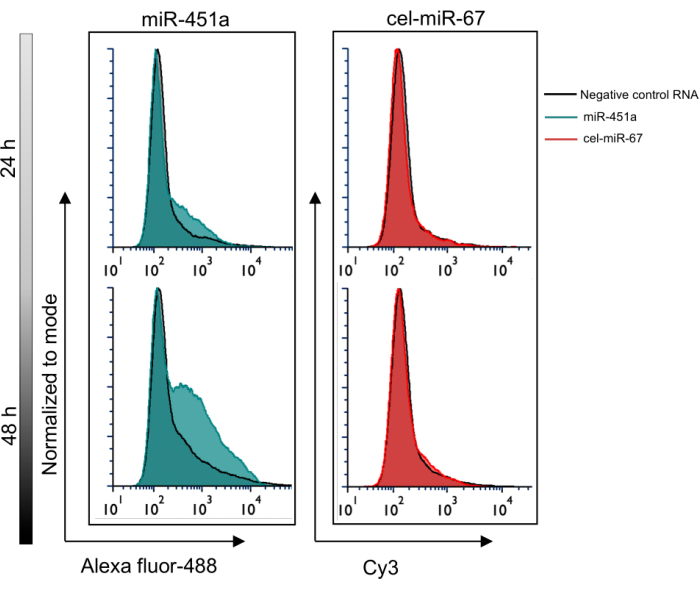
Figure 4: Signal detection in EVs through flow cytometry. Fluorescence corresponding to EV-miR-451a and cell-cel-miR-67 was detected in EVs 24 h and 48 h post-transfection. The signal corresponding to EV-miR-451a (Alexa fluor-488) was captured in EVs 48 h following transfection. In contrast, the signal for cel-miR-67 was not detected in EVs. A blank PBS sample was used to set up the gating for this experiment, as shown in Figure 2B. EV samples isolated from cells transfected with non-fluorescent scrambled RNA were used as a negative control. Please click here to view a larger version of this figure.
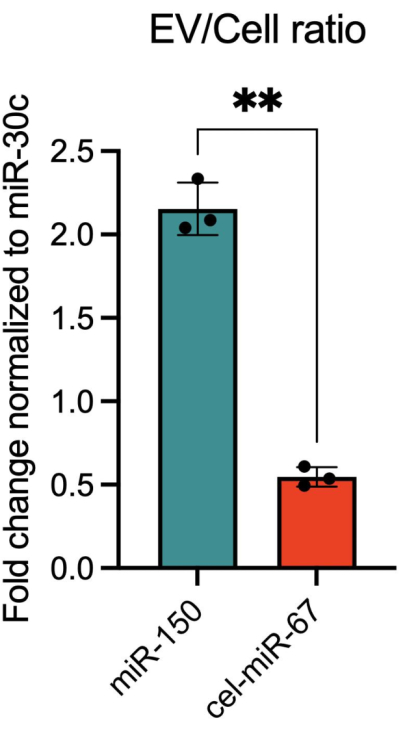
Figure 5: Quantification of EV-miRNA and cell-miRNA in cells and EVs using qRT-PCR. Calu6 cells were transfected with 2 nM of miR-150 and cel-miR-67 each. EVs were collected 48 h after transfection. EV enrichment was calculated as the ratio of EV expression divided by cellular expression in transfected Calu6 cells for miR-150 and cel-miR-67. Based upon RNA sequencing results comparing the expression of miRNAs in Calu6 cells and their corresponding EVs, miR-30c was used as an endogenous control and a normalizer for fold change computation. The data presented are from one biological replicate, with each data point presenting triplicate qRT-PCR reactions. Data are expressed as mean ± SD, and p-values were computed using an unpaired t-test with Welch's correction (**p < 0.01). Please click here to view a larger version of this figure.
| Strengths | Weaknesses |
| Allows for simultaneous assessment of selective export of multiple miRNAs in cells and respective EVs | Require additional procedures to differentiate between active and passive export phenomenon |
| Assessment of crude heterogenous population is possible and does not require prior separation of EV populations. Can be used to analyze different types of EVs NOTE: Threshold for detection of different types of particles will vary |
Low sensitivity does not allow for detection of limited concentrations of miRNAs in cells or EVs. This can be overcome by incorporating downstream qRT-PCR analysis following RNA isolation from EVs |
| Requires a short amount of time for set up and analysis in comparison to standard methods used for miRNA quantification | Lack of standardization protocols for different types of equipment, cell-lines, and various fluorophores. |
Table 1: The major strengths and weaknesses of the protocol.
