Immediately following isolation of LMMP-derived cells, neurons and other cell types will not be readily evident. Living, round cells of indistinct phenotype can be seen as well as tissue detritus from incompletely digested tissue fragments and connective tissue. This flotsam is of no concern and will be largely removed with the first media change in two days. Do not attempt to clean the slides before this as the healthy, viable cells will be removed as well.
After one day in culture, neurons will begin to show neurite outgrowth. Specific identification of neurons may still be indistinct at this time. Provided the cells are adherent enough to transfer to an experimental chamber, they will be ready for electrophysiological study after one day of culture. However, the cells are more adherent after two days in culture and ideal for function studies (electrophysiology, calcium imaging) from approximately days 2 – 5.
Morphological features of neuron become distinct after approximately a week in culture (Figure 1). Ideal immunocyctochemical features can be identified after about ten days, when neurons display long projections and grow in an almost ‘ganglionic’ like fashion interspersed with glia (Figure 2). This staining suggests it may be possible to study the synaptic interactions of these cells using this methodology.
After one day in culture, neurons are recognized by their sine que non or ‘defining feature’, the action potential (Figure 3). Electrophysiologically, they can be functionally classified into at least two neuronal types: neurons that contain an after-hyperpolarization and increased current/density relationships of sodium and potassium and neurons that do not have an after-hyperpolarization and significantly less sodium and potassium conductance (Figure 4). AHP positive and negative neurons appear to correlate with AH and S neurons seen in previous guinea work, respectively. AHP positive neurons (AH neurons) have multiple long projections originating from around the cell body, while the AHP negative neurons (S neurons) have one long projection which branches many times. This, in conjunction with the immunocytochemical coding in Figure 1, suggests that neuronal population is very heterogeneous.
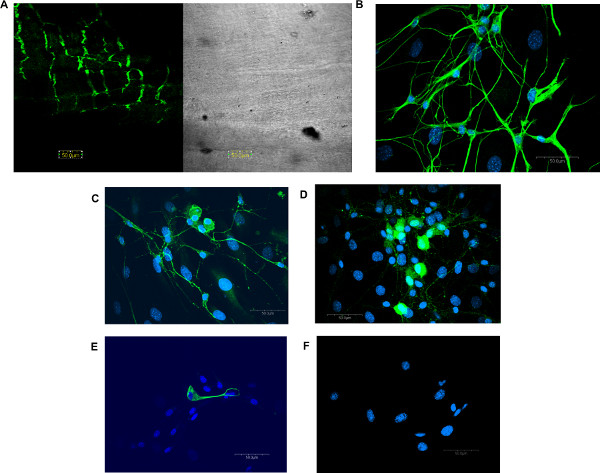
Figure 1. Immunohistochemical characterization of enteric neurons and glia isolated from the mouse longitudinal muscle. Confocal microscopy revealed neuronal-specific β-III-tubulin (Abcam, rabbit, 1:1,000) staining in whole mount ileal longitudinal muscle (A) preparation from the mouse. Cells isolated from longitudinal muscle/myenteric plexus (LMMP) preparations contain neurons (B; β-III-tubulin, Abcam, rabbit, 1:1,000) that stain positively for calbindin (C) (Chemicon, rabbit, 1:1,000) and calretinin (D) (Swant, rabbit, 1:2,000). Glia cells (E) were visualized with the glia-specific marker GFAP (Chemicon, mouse, 1:500). Antibodies were visualized via appropriate goat secondary antibody Alexa 488 (green, Molecular Probes, 1:1,000)0. Nuclei were visualized using Hoescht 33342 (blue, C-G, 1 μg/ml). No staining was seen when primary antibody was omitted (F). Modified and reprinted from Smith, T.H., et al. Morphine Decreases Enteric Neuron Excitability via Inhibition of Sodium Channels. PLoS One., doi:10.1371/journal.pone.0045251.g001 (2012). Click here to view larger figure.
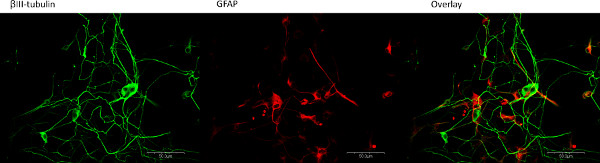
Figure 2. Neurons and glia isolated from the mouse longitudinal muscle grow in close proximity to one another. Confocal microscopy images indicate the neurons (green, β-III-tubulin, Abcam, rabbit, 1:1,000) and glia (red, GFAP, Chemicon, mouse, 1:500) readily grow adjacent to one another and appear to interact in vitro. Click here to view larger figure.
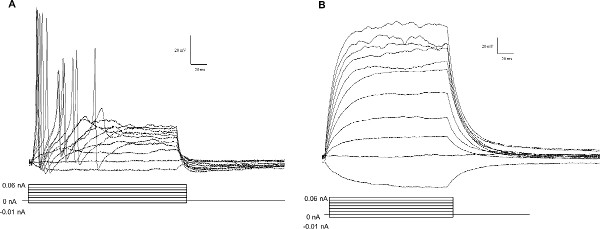
Figure 3. Electrophysiology of cultured enteric neurons and glia. In current clamp mode, all neurons (A) displayed action potentials upon current injection. Glia (B) do not have action potentials but do display large electrotonic potentials in response to current injection. Protocols in A & B start with a current injection of -0.01 nA and increase to 0.09 nA in eleven 0.01 nA steps. Click here to view larger figure.
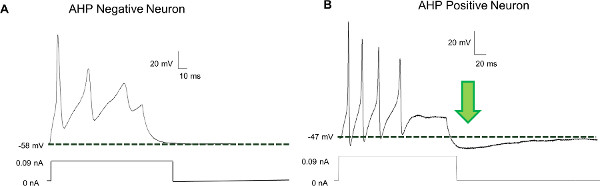
Figure 4. Neurons cultured from the mouse ileum are an electrophysiologically heterogeneous population. In current clamp mode, a current injection of 0.09 nA into neurons results in action potentials. S neurons (A), immediately return to the resting membrane potential following stimulation. AH-type neurons (B) display an afterhyperpolarization (AHP) following stimulation in which the resting membrane potential falls below baseline before slowly returning to the initial value. Modified and reprinted from Smith, T.H., et al. Morphine Decreases Enteric Neuron Excitability via Inhibition of Sodium Channels. PLoS One., doi:10.1371/journal.pone.0045251.g001 (2012).
