Two-color infrared fluorescence detects both strong and weak bands on the same membrane with enhanced sensitivity and the highest signal-to-noise ratio to produce clear Western blot images. Typical results generated by two-color Western blot detection visualized in both the 700 nm and 800 nm fluorescent channels are exemplified in Figures 1 and 2. The uppermost Western blot in Figure 1 shows the concurrent (overlay) detection of total ERK1/2 in the 800 nm channel (green) and β-Actin in the 700 nm channel (red) of two-fold serial dilutions of lysates from the human-derived melanoma cell line, WM793. In addition, this membrane has also been cut into sections based on the molecular weights of the desired protein targets in order to allow for more than two antibodies to be examined on the same blot. For instance, the membrane analyzed in Figure 1 was cut in half in order to probe the lower portion of the membrane with a third antibody, total BIM (700 nm channel-red) without inferring with the detection of both total ERK1/2 and β-Actin. The first panel of Figure 2 further demonstrates the two-color Western blot fluorescent detection of phospho-ERK1/2 (800 nm channel-green) and total ERK1/2 (700 nm channel-red) with overlapping phospho-ERK and total ERK signals displayed in yellow of WM793 melanoma cells treated in the presence and absence of the MEK1/2 inhibitor, PD0325901 (MEKi), which suppresses the ERK1/2 MAP kinase signaling pathway. Moreover, the 700 nm and 800 nm fluorescent images can also be converted to the standard black and white Western blot images for each antibody (Figures 1 and 2). In general, the majority of Western blot images produced by this protocol will result in high-quality images as exhibited in Figures 1 and 2. However, depending on the selectivity, specificity, and quality of the primary antibody, less than optimal images can result in a small portion of blots analyzed by this methodology. For example, as observed in the bottom two panels of Figure 2, the subpar quality of the phospho-BIM antibody in the WM793 melanoma cells treated with and without MEK inhibition resulted in a Western blot image with faint protein bands and extremely high membrane background.
Simultaneous infrared fluorescent imaging of two targets can also provide the opportunity for quantitative analysis of Western blots over a broad, linear dynamic range by efficiently and accurately detecting both strong and weak bands on the same membrane. Quantification of protein bands are calculated as an integrated intensity, which is proportional to the amount of fluorescent-labeled antibodies on the membrane and is independent of both size of the shape drawn around the band and resolution. This is based on the following criteria: 1) area of the shape; 2) number of pixels enclosed in the shape; 3) average intensity of the pixels selected as the background and; 4) total intensity of the pixels enclosed in the given shape10. Figure 3A shows the protein quantification of the Western blots illustrated in Figure 1. Quantification of total ERK1/2, β-Actin, and total BIM exhibit a linear relationship between an increase in the integrated intensity of the fluorescent signal and the increase in protein concentration (Figure 3A). Moreover, Figure 3B is another example of how to quantify protein levels from fluorescent Western blots, which shows the protein quantification of phospho-ERK1/2 normalized to total ERK1/2 protein levels of WM793 melanoma cells treated with and without MEK inhibition from Figure 2. From this quantification, the phosphorylated ERK1/2 levels can be calculated as a percent of the control levels. In this case, phospho-ERK levels of melanoma cells treated with the MEK inhibitor were 0.6% of the control.
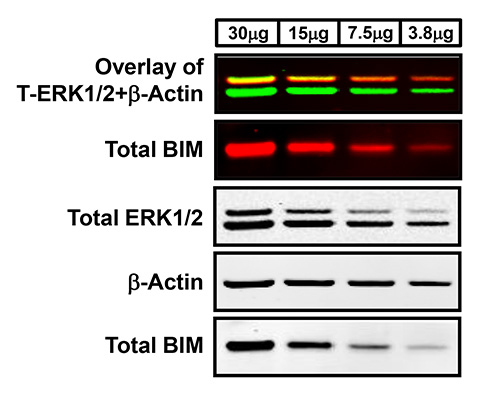
Figure 1. Western blot analysis using two-color infrared fluorescent protein detection. Lysates of two-fold serial dilutions of WM793 human-derived melanoma protein were separated using a NuPAGE Novex Bis-Tris gel with protein transferred onto PVDF membrane using the iBlot transfer apparatus. Membranes were blocked in Odyssey Blocking Buffer and probed with the following primary antibodies: rabbit anti-total ERK1/2, rabbit anti-total BIM, and mouse anti-β-Actin. Antigen-antibody complexes were detected using fluorescent goat anti-rabbit IRDye 800 (green) or 680 (red) and goat anti-mouse IRDye 680 (red) secondary antibodies, respectively, and visualized with the LI-COR Odyssey Classic Infrared Imaging System. The first Western blot panel is the overlay of the simultaneous detection of total ERK1/2 (800 nm channel-green) and β-Actin (700 nm channel-red) fluorescent signals. The second panel is the single-channel fluorescent detection of total BIM on the same membrane that was separated using a razor blade based on the molecular weights of these target proteins. The lower three Western blot panels are the black and white images of the separated single-channel fluorescent images.
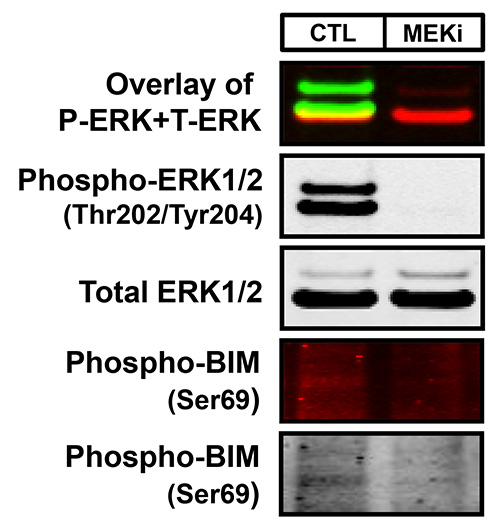
Figure 2. An example of both a high- and poor-quality Western blot image using infrared fluorescence. WM793 melanoma cells treated with either DMSO (CTL) or 2 μM of the MEK inhibitor, PD0325901 (MEKi), for 18 hr were analyzed as described in Figure 1. The membrane was probed with rabbit anti-phospho-ERK1/2 (Thr202/Tyr204), mouse anti-total ERK1/2, and mouse anti-phospho-BIM (Ser69) with antigen-antibody complexes detected with goat anti-rabbit IRDye 800 (green) and goat anti-mouse IRDye 680 (red). The first Western blot panel is the overlay of the simultaneous detection of both phospho-ERK1/2 (800 nm channel-green) and total ERK1/2 (700 nm channel-red) fluorescent signals. The second and third panels are the black and white images of the single-channel fluorescent images of phospho and total ERK1/2. The bottom two panels are the fluorescent and black and white images of phospho-BIM. These images represent an example of a subpar Western blot that can result with this method due to the substandard quality of the primary antibody.
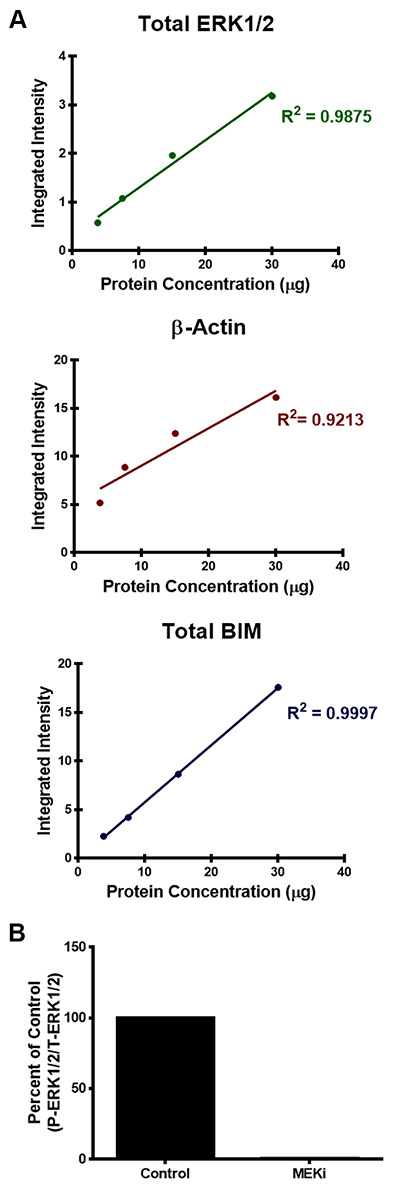
Figure 3. Examples of protein quantification of the infrared fluorescent Western blots. A) Quantification of total ERK1/2, β-Actin, and total BIM protein levels was determined by calculating the integrated intensity of each protein band. The integrated intensity is proportional to the amount of fluorescent-labeled secondary antibodies on the membrane. R-squared values were also used to evaluate the linear regression for each of the antibodies analyzed in Figure 1. B) Quantification of phospho-ERK1/2 normalized to total ERK1/2 protein levels from Figure 2 was calculated using the integrated intensity of the fluorescent signal and data is presented as a percent of the DMSO-control.
