The top panel of Figure 3 displays examples of the calculated mass flux of VX and HD from SD-painted substrates based on time-resolved mass spectrometry for the main mass fragments of VX and HD (mass-to-charge ratio, m/z = 114 and 109, respectively). A quadrupole mass spectrometer has three main components: an ionizer, a mass analyzer or filter, and a charge detector. Gas species are ionized via electron impact ionization (hot filament style electron source), and the produced ions are injected into a quadrupolar mass filter en route to reaching a Faraday cup or electron multiplier detector for measuring ion current (proportional to partial pressure of measured gases). For instrument used in this method, the ionizer was located in close (<3.5 cm), line-of-sight proximity to the substrate surface so that species emitted from a contaminated substrate (emission angle peaked at substrate normal) were detected preferentially and pseudo-instantaneously. The ions that comprise the ion current at the detector are assigned to mass fragments (m) with a specific ionization state (z) that results from the electron impact ionization process through the use of independent mass spectrometry standards and databases (e.g., NIST mass spectra database). The relative abundance of particular ions created in an electron impact ionization event is molecule-specific, and great care is needed in assigning mass spectral signatures especially for larger molecules, such as VX and HD, which rarely undergo single ionization or fragmentation events. Before time-resolved experiments were performed, full mass spectra for equivalent substrate preparation conditions were recorded to verify the presence and relative abundance of specific m/z peaks for the species introduced into the analysis chamber. This verification included confirming that the intensity of the expected main mass fragment for the analyte of interest was not convolved with contributions from other species in the system. The time-resolved measurements in subsequent experiments then recorded the intensity of specific m/z peaks as a function of time associated with the analytes of interest.
Raw mass spectrometry data are in the form of partial pressures of gases as measured at the ionizer of the spectrometer. For this study, the partial pressure values for the primary HD and VX fragments used for the flux calculations are based on a nitrogen ionization cross section standard with no correction for possible molecule-specific fragmentation efficiency. As specified in the protocol, Step 6.1, these partial pressure measurements at the detector are first converted to a mass flux using the Hertz-Knudsen formula that relates:
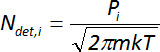 Equation 1
Equation 1
where Ndet,i, is incident molecular flux, Pi, is measured partial pressure (Pa), m is the molecular mass of the species (g/molecule), k is the Boltzmann constant (J/K), and T is the chamber temperature (K). Then, in order to determine the flux from the contaminated surface, Nc, the mass flux measured at the detector, Ndet, i, is multiplied by the ratio of the mass spectrometer detector area (cross sectional area of the detector parallel to the contaminated surface) to the area of contamination on the surface:
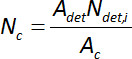 Equation 2
Equation 2
where Adet is the area of detector and Ac is the contaminated area on the substrate. The data shown in the top panel of Figure 3 focus on the time regime associated with diffusive mass transport for species originating from the subsurface of the substrate. Measurements prior to mass spectrometry demonstrated that both VX and HD drops did not remain sessile on the SD-paint with residual bulk liquid on the substrate prior to introduction to vacuum; the average final contaminated area for VX and HD was 2.6 ± 0.1 and 5.9 ± 0.6 cm2, respectively. Early time regime (<1,000 sec) data are not included in these plots, which are partly influenced by mass spectrometer equilibration for ionizer operation at relatively high partial pressures as well as desorption of bulk liquid contaminant that is trapped or weakly bound at the surface of the substrate. The data demonstrate a diffusion-limited regime that begins with an exponential decrease in mass flux rate and is the source of the data used for parameter estimation of diffusive transport from the subsurface of the contaminated substrate. The definition of the transition in regimes was confirmed by measuring evaporation of VX and HD liquid deposited on borosilicate glass substrates (impermeable material), which demonstrated a constant evaporation rate for both VX and HD until the deposited liquid was depleted. For the purposes of illustrating the various interactions that comprise the complex system under study, the bottom panel in Figure 3 shows the background signals measured from an uncontaminated paint substrate. Note the non-monotonic time evolution as well as the coincidence of features across species profiles. As measurements are recorded under vacuum conditions, any of the species trapped in the paint substrate are capable of diffusing out of the coating. This observation illustrates the possibility that the rate of mass flux out of the substrate (proportional to partial pressure) can be influenced by the relative abundance of rate of depletion of other species in the coating, including water and paint solvents.
Parameter estimation was performed using the diffusion-limited vapor emission flux. The calculated fit, which is an analytical solution to Fick’s second law of diffusion, allows for the determination of saturation and diffusivity constant for the specific contaminant-material combinations8. Vapor emission, including the mass flux magnitude as well as the time rate of change of the mass flux, from a material is driven by the contaminant distribution with respect to the material as well as the transport parameters associated with the contaminant in the material9-11. If the distribution of a contaminant is known, then the contaminant flux from the material can be predicted1. In contrast, if the vapor emission flux is measured (similar to the experimental procedure described herein), the concentration distribution in the substrate and transport parameters may be determined; a technique that is commonly referred to as inverse analysis12,13. In this experiment, an expression for vapor flux from the substrate can be derived via a one-dimensional (1D) analytical solution to Fick’s second law for the contaminant concentration profile in a finite thickness coating, assuming diffusive mass transport4,13. Figure 4 illustrates the different boundary conditions associated with constraining the analytical solution for molecular diffusion in a finite thickness absorbing coating. The full details of the calculation are documented elsewhere4, and the results are summarized in Table 1.
The mass transport parameters estimated from the vapor flux measurements were implemented to predict the contaminant concentration distribution and evolution within the SD paint coating. This result included representing the concentration field over the thickness of the paint at the start of diffusion of the contaminant out of the paint coating (after bulk liquid evaporation from the surface). The top panel of Figure 5 illustrates simulation results for the spatial-dependent concentration distribution of HD or VX in the SD paint coating. Although the coating absorbed a higher mass of HD, as indicated by a greater saturation concentration listed in Table 1, it is more localized near the surface of the substrate compared with VX. The bottom panel of Figure 5 illustrates the resulting vapor flux of the contaminants from the paints, which could yield health hazards to personnel in the vicinity of the material. The differences in transport phenomena between HD and VX demonstrate that intermolecular interactions can alter the transport of absorbing molecules through the paint systems to generate different distributions.
Figure 1. Substrate preparation and measurement flow chart that illustrates steps for preconditioning, contaminating, aging, and measuring subsequent emission of agent from substrates. Please click here to view a larger version of this figure.
Figure 2. Schematic of the high vacuum vapor emission chamber. Substrates are loaded onto a temperature-controlled substrate holder that directly faces a quadrupole mass spectrometer. The chamber has a base pressure (no substrate loaded) of <10−7 Pa and is pumped by a turbo molecular drag pump that is supported by a diaphragm backing pump. Substrates are introduced with the vacuum chamber vented, and measurements of mass flux at the mass spectrometer commence once the system is pumped down <10−2 Pa. Substrate diameter serves as a scale reference. Please click here to view a larger version of this figure.
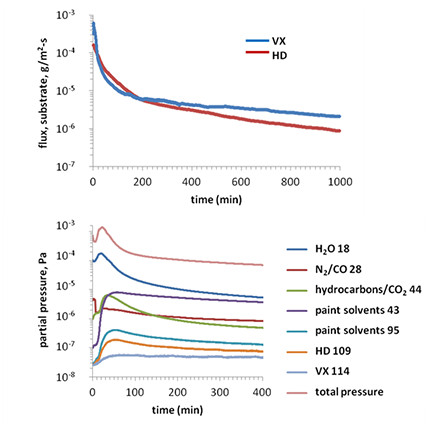
Figure 3. Top: VX (blue) and HD (red) calculated flux from substrates based on experimental measurement of vapor emission from contaminated SD chemical agent resistant coating substrates. The partial pressure (derived from ion current) measured at the mass spectrometer of the main fragments for a given analyte (VX: m/z = 114; HD: m/z = 109) can be related directly to the original mass flux from the substrate, assuming line-of-sight detection of species emitted from the material. Data shown are for the diffusion-limited mass transport regime only when bulk, surface-bound contaminant liquid has desorbed. Bottom: Background partial pressure measurements for gas species emitted by an uncontaminated paint substrate. For the purpose of illustrating the complexity of the system under study, the full time-resolved profiles are included from the initial point of collecting data (when total chamber pressure dropped below 10−2 Pa, the earliest point at which the mass spectrometer could record). The color key illustrates specific m/z values that correspond to the identified detected species.
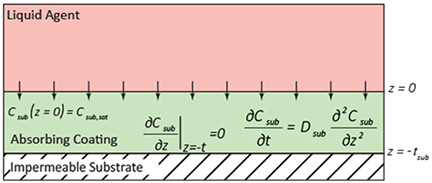
Figure 4. Definition of 1D molecular diffusive absorption governed by Fick’s law, including boundary conditions. Definition of variables: Csub, concentration of contaminant in the substrate; Csub,sat, saturation concentration; z, distance along the thickness of the absorbing coating; Dsub, diffusivity of contaminant in the coating; tsub, thickness of the paint coating. For the model that describes the measurement under vacuum for contaminant evolution from the substrate, the model assumes that at z = 0, there will be a vacuum-coating interface, the point at which diffusion-limited transport dominates. The form of the analytical solution to Fick’s law with these boundary conditions and its subsequent use with inverse analysis calculations are detailed in reference 4.
Figure 5. Top: Calculated concentration profiles from experimentally determined mass flux from paint substrates for each agent as a function of paint coating depth using inverse analysis. The profiles reflect the distribution at the point where the vapor flux from the contaminated material come solely from the subsurface and is determined by diffusive mass transport. Overall, in this case, more HD mass is absorbed and does so in a thinner portion of the paint film compared to VX. This difference is reflected in the calculated saturation concentration and diffusion coefficient values. Bottom: Calculated vapor flux profiles based on the starting point concentration profiles in the top portion of the figure. The calculated, time-dependent vapor flux matches the experimental data shown in Figure 3 with regard to predicting the relative difference in evolution of each agent. Please click here to view a larger version of this figure.
| Agent | Diffusivity (m2/s) | Saturation (g/m3) |
| VX | 9.91 ± 0.07 × 10–14 | 5.32 ± 0.03 × 104 |
| HD | 2.11 ± 0.04 × 10–14 | 1.17 ± 0.01 × 105 |
Table 1. Mass transport parameter estimation results for VX and HD interacting with SD paint. Table adapted from reference 4.
