This protocol details a method to reconstitute chorophyll a/b binding proteins in vitro. This technique permits the folding of these pigment-protein complexes in vitro starting from the apoprotein, which can be obtained by overexpression in a heterologous system, and pigments extracted from plant or algae. After reconstitution, the refolded pigment-protein complex is purified from the excess of pigments and the unfolded apoprotein in two steps. The first step (Figure 1 A-B) is based on the presence of His-tag at the C-terminal of the protein, which permits the removal of large part of the unbound pigments. The second purification step utilizes sucrose density gradient centrifugation, (Figure 2) where the unfolded protein usually migrates slower than the green band containing the reconstituted protein. The goal of the reconstitution in vitro is to obtain complexes with the same properties as the native ones. To illustrate this outcome, the spectroscopic properties of an in vivo light-harvesting complex is compared with the same LHC complex reconstituted in vitro13,20,21. The absorption spectrum of the LHCs in the visible range (350 nm and 750 nm) depends on the pigment composition of the complex, as well as on the pigment’s environment (which includes the protein) and it is thus a sensitive tool to check the quality of the reconstitution. In Figure 3, the absorption spectrum of CP24, a chlorophyll a/b binding protein from Arabidopsis thaliana, reconstituted in vitro, is compared with the spectrum of the same complex purified from Arabidopsis thylakoids21. In the spectra, it is possible to recognize the Qy and the Soret transition of Chl a (peaks at 671/439 nm) and Chl b (peaks at 649/466 nm). The native and reconstituted complexes show identical absorption spectra, indicating a virtually identical pigment composition and organization. Fluorescence spectroscopy can be used to assess the quality of the reconstituted complex. The fluorescence emission spectra is measured upon excitation at different wavelengths, which excite preferentially different pigments: Chl a at 440 nm, Chl b at 475 nm, and Xanthophylls at 500 nm. In a properly folded protein-pigment complex, Chl b and Xanthophylls transfer their excitation energy primarily to Chl a within a few picoseconds, and the fluorescence originates from a thermally equilibrated system resulting in a single peak with the same shape and maxima at all three excitation wavelengths (Figure 4A–B). The presence of Chl b not coordinated to the protein can be recognized by an additional peak or shoulder around 650 nm upon 475 nm excitation (Figure 4C). The presence of free Chl a instead leads to additional emission around 675 nm, which is mainly present upon 440 nm excitation. The fluorescence emission spectra upon 475 nm excitation of both reconstituted and the native CP24 complexes (Figure 4D) show a single peak at 681 nm, indicating that reconstituted complex is correctly folded. An additional confirmation that the pigment-protein complex is correctly reconstituted comes from circular dichroism (CD) measurements. The CD signal in the visible region depends on the excitonic interactions between pigments and it is thus very sensitive to even small changes in the organization of the chromophores22. Figure 5 shows the CD spectra of reconstituted and native CP24, with the typical fingerprint peaks at 681 nm, 650 nm and 481 nm. In conclusion, the high similarity between the spectroscopic properties of native and the reconstituted CP24 confirms that the reconstitution procedure yields native-like complexes suitable for in vitro study of light-harvesting proteins.
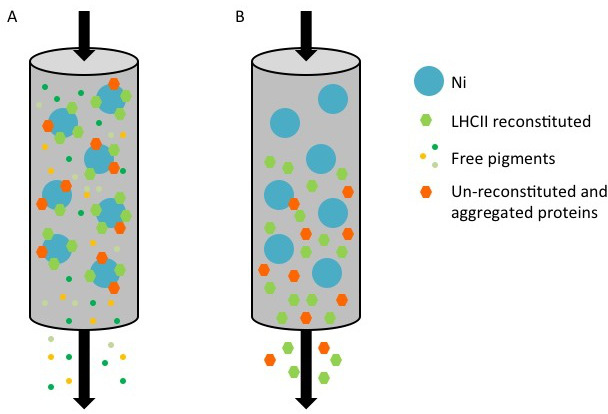
Figure 1. Representation of the purification of recombinant LHC proteins with a His tag using a nickel column. (A) During the purification, His-tagged protein, comprised of both reconstituted complexes (green hexagon) and un-reconstituted/aggregated protein (orange hexagon) are bound to the surface of the Ni-Sepharose (blue spot), while unbound pigments (small colored spots) flow through. (B) When the column is washed with the elution buffer containing imidazole, the reconstituted and un-reconstituted proteins are collected in the flow through.

Figure 2. Sucrose gradient of reconstituted LHCII after purification by nickel column. The reconstituted complexes are separated from the free pigment by the density gradient. The dark green band represents reconstituted LHCII and the pale green background is composed of free pigments.
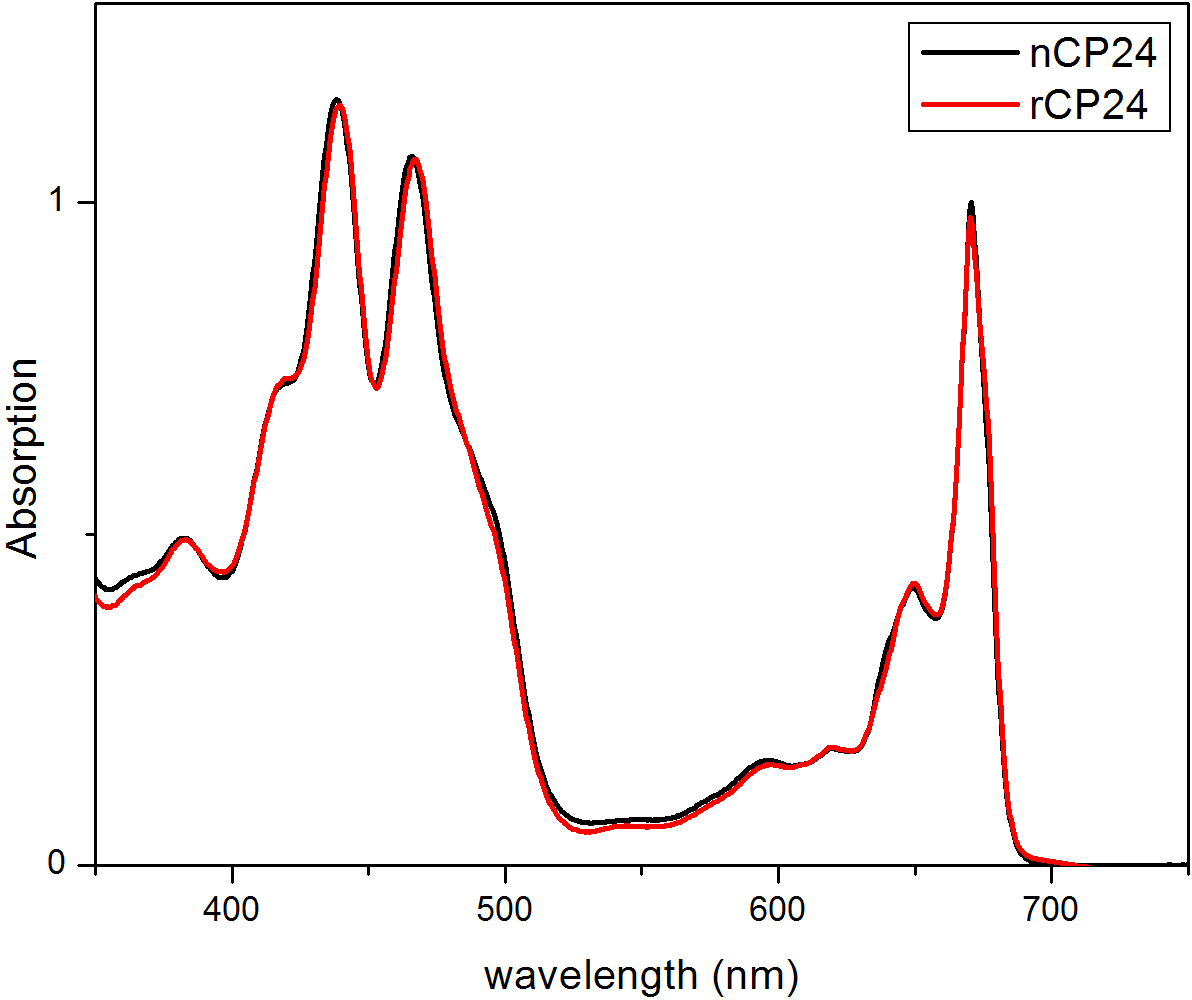
Figure 3. Absorption spectra of reconstituted protein CP24 (rCP24, red line) and the native one (nCP24, black line) isolated from Arabidopsis thaliana. In both spectra, it is possible to recognize the Qy and the Soret transition of Chl a (peaks at 671/439 nm) and Chl b (peaks at 649/466 nm). This figure has been modified from Passarini et al. 201421.

Figure 4. Fluorescence emission spectra. The fluorescence emission spectra of reconstituted CP24 wildtype complex (A) and normalized to the maximum (B) showing efficient energy transfer from Chl b and Xanthophyls to Chl a. (C) Fluorescence emission spectra of reconstituted CP24 (rCP24) and the native complex (nCP24) isolated from Arabidopsis thaliana. The spectra are normalized to the maximum of the peak (D). Please click here to view a larger version of this figure.
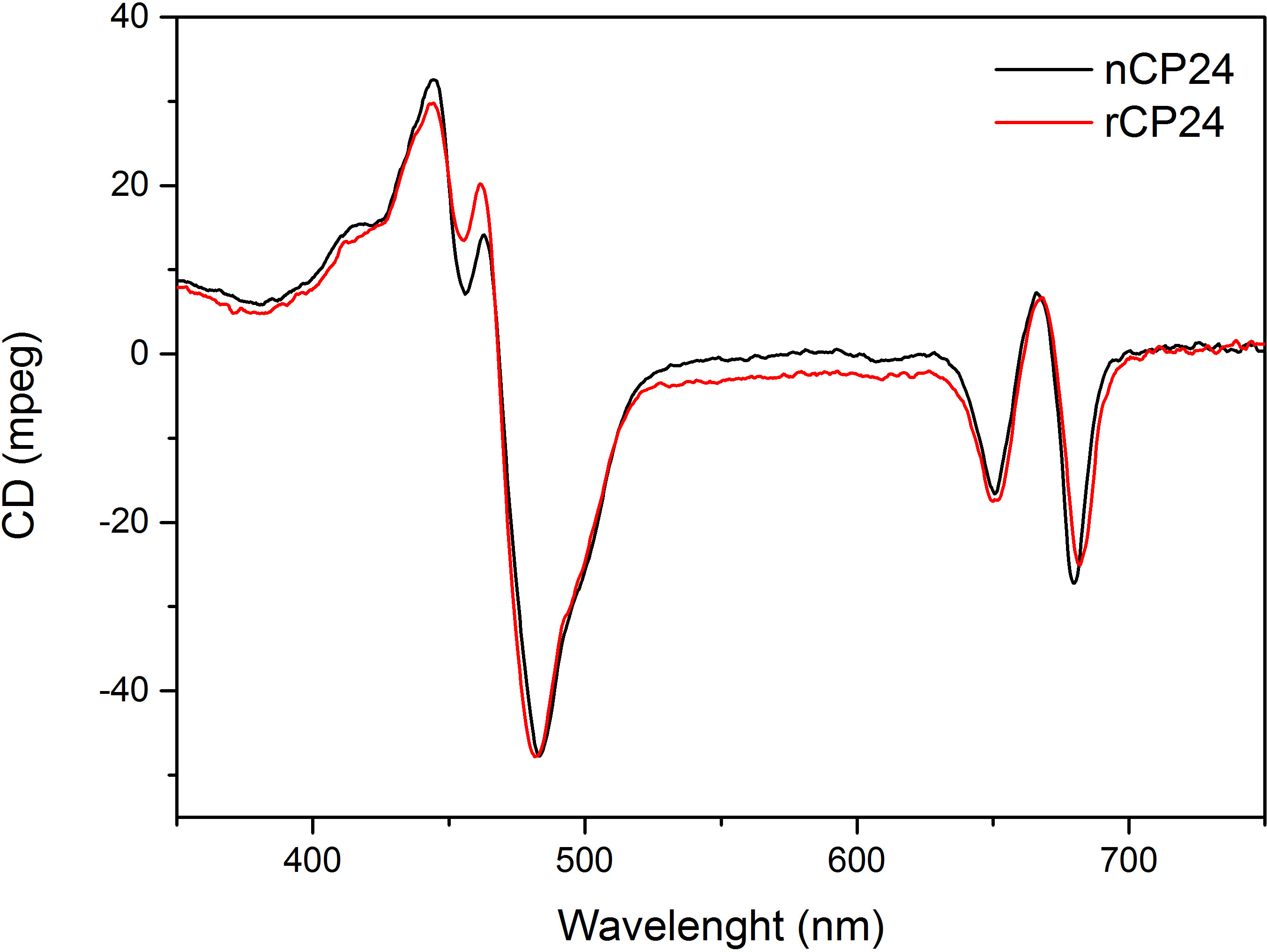
Figure 5. Circular Dichroism Spectra. Reconstituted CP24 (rCP24, red line) and the native complex (nCP24, black line) isolated from Arabidopsis thaliana shows very similar spectra.
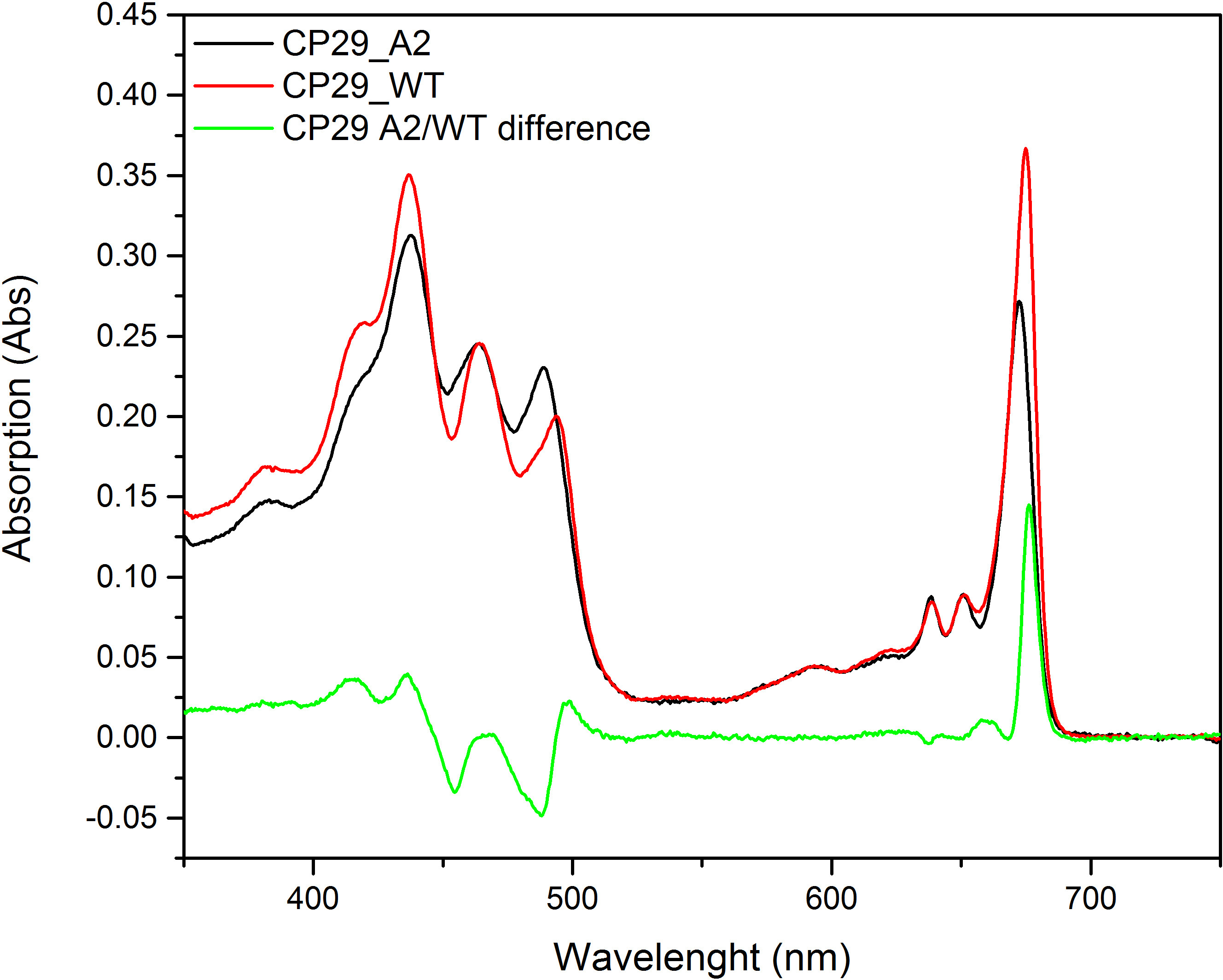
Figure 6. Absorption spectra of CP29 wild type (CP29_WT) and mutated CP29 (CP29_A2). The green line shows the differences between the two plots.
| All the buffers can be stored at 4 °C. | |||
| Components | Final Concentration | Additional notes | |
| Grinding Buffer | Sorbitol | 0.4 M | |
| Tricine | 0.1 M | pH 7.8 | |
| NaCl | 10 mM | ||
| MgCl2 | 5 mM | ||
| Milk Powder | 0.5% w/v | ||
| Wash Buffer | Sorbitol | 50 mM | |
| Tricine | 5 mM | pH 7.8 | |
| EDTA | 10 mM | pH 8 | |
| Lysis Buffer | Tris | 50 mM | pH 8 |
| Sucrose | 2.5% w/v | ||
| EDTA | 1 mM | pH 8 | |
| Detergent buffer | NaCl | 200 mM NaCl | |
| Deoxycholic acid | 1% w/v | ||
| NONIDET P-40 | 1% w/v | ||
| Tris | 20 mM | pH 7.5 | |
| EDTA | 2 mM | pH 8 | |
| beta-mercaptoethanol | 10 mM | ||
| Triton Buffer | Triton X-100 | 0.5% w/v | |
| Tris | 20 mM | pH 7.5 | |
| beta-mercaptoethanol | 1 mM | ||
| Buffer TE | Tris | 50 mM | pH 8 |
| EDTA | 1 mM | pH 8 | |
| Reconstitution Buffer | HEPES | 200 mM | |
| Sucrose | 5% w/v | ||
| Lithiumdodecylsulfate (LDS) | 4% w/v | ||
| Benzamidine | 2 mM | ||
| Aminocaproic Acid | 10 mM | ||
| OG Buffer | Octylglucoside | 1% w/v | |
| Sucrose | 12.5% w/v | ||
| NaCl | 0.2 M | ||
| HEPES | 20 mM | ||
| Imidazole | 10 mM | ||
| OG Rinse Buffer | n-Dodecyl-beta-D-Maltoside (β-DM) | 0.06% w/v | |
| HEPES | 40 mM | pH 7.5-9 | |
| NaCl | 0.2 M | ||
| Elution Buffer | Imidazole | 0.5 M | |
| n-Dodecyl-beta-D-Maltoside (β-DM) | 0.06% w/v | ||
| HEPES | 40 mM | pH 8 | |
| NaCl | 0.2 M | ||
| Sucrose Solution | Sucrose | 20% w/v | |
| n-Dodecyl-beta-D-Maltoside (β-DM) | 0.06% w/v | ||
| HEPES | 0.01 M | pH 7.6 | |
| Acetone 80% buffered with Sodium Carbonate | Acetone | 80% v/v | |
| Sodium Carbonate | 1 M | ||
| Ethanol 96% buffered with Sodium Carbonate | Ethanol | 96% v/v | |
| Sodium Carbonate | 1 M | ||
Table 1. List of buffers and solutions used in this protocol.
| Chla a/b mix | Chla a/b | Chl a | Chl b | Neo | Viola | Lute | Chl tot | Chl/Car | |
| nCP26 | – | 2.2±0.05 | 6.2 | 2.8 | 0.61 | 0.38 | 1.02 | 9 | 4.5±0.1 |
| rCP26 | 8 | 2.71±0.05 | 6.57 | 2.43 | 0.72 | 0.32 | 0.97 | 9 | 3.9±0.04 |
| rCP26 | 5.5 | 2.25±0.05 | 6.23 | 2.77 | 0.77 | 0.3 | 0.96 | 9 | 4.0±0.1 |
| rCP26 | 3 | 2.08±0.04 | 6.08 | 2.92 | 0.76 | 0.3 | 1.04 | 9 | 4.1±0.1 |
| rCP26 | 1 | 1.7±0.05 | 5.7 | 3.3 | 0.7 | 0.3 | 0.9 | 9 | 4.3±0.05 |
| rCP26 | 0.3 | 1.11±0.04 | 4.7 | 4.28 | 0.7 | 0.3 | 0.9 | 9 | 4.2±0.2 |
| rCP26 | 0.05 | 0.23±0.01 | 1.4 | 5.6 | 0.58 | 0.24 | 1.11 | 7 | 3.1±0.06 |
| rCP26 | <0.01 | 0.11±0.01 | 0.7 | 6.3 | 0.64 | 0.3 | 1.08 | 7 | 3.06±0.06 |
Table 2. Pigment content of CP26 native complex compared to reconstituted protein-pigment complexes with different Chl a/b Ratios39.
