1. Yeast Growth Assay to Identify Candidate Mutants Defective in Protein Degradation
- Transform wild-type and mutant yeast cells with a plasmid encoding an unstable protein fused, in frame, to a reporter metabolic enzyme.
- Inoculate transformants in 5 ml of synthetic defined (SD) minimal medium that is selective for cells harboring plasmid molecules. Incubate overnight at 30 °C, rotating.
- Measure the optical density at 600 nm (OD600) of each overnight culture.
NOTE: Following overnight incubation, cells in culture may be in either logarithmic or stationary growth phase but should have reached a minimal OD600 of 0.2. Very slow-growing yeast strains may require incubation times longer than one night, or inoculation of a greater number of cells, as determined empirically. - Prepare six-fold serial dilutions of transformed yeast cells in a sterile 96-well plate, beginning with cells diluted to an OD600 of 0.2. Place each yeast transformant to be assayed in a different row in the 96-well plate.
- For each transformant, calculate the volume of overnight culture required to dilute cells to an OD600 of 0.2 in a final volume of 200 µl. Add this volume of overnight culture to the corresponding well in Column 1. Add the appropriate amount of sterile water to bring the volume to 200 µl.
- For each row of yeast, add 125 µl of sterile water to the wells in Columns 2, 3, and 4.
NOTE: Individually wrapped sterile 96-well plates may be packaged with sterile lids. The lids may be used as reservoirs for the sterile water that is distributed in this step. This allows simultaneous transfer of sterile water to all wells in a given column with a multichannel pipettor. - Mix the contents of the first column (yeast diluted to an OD600 of 0.2) by pipetting up and down with a multichannel pipettor.
- Transfer 25 µl of yeast from Column 1 to Column 2, using a multichannel pipettor. Mix by pipetting up and down. Transfer 25 µl from Column 2 to Column 3, and 25 µl from Column 3 to Column 4 (mixing well at each step).
- Mix each sample with a multichannel pipettor. Proceeding from most dilute to least dilute columns of yeast, pipette 4 µl of each sample onto two plates containing the appropriate selective medium. Use one plate with medium that maintains plasmid selection (this plate serves as a yeast spotting and growth control). Use a second plate with medium that selects for plasmid maintenance and expression of the unstable protein fused to the reporter enzyme. Because yeast settle rapidly, mix cells by pipetting up and down at regular intervals.
NOTE: Drier plates will more readily absorb liquid than freshly prepared plates and are therefore recommended for these experiments. Damp plates may be dried by incubation at room temperature in low humidity for 1 – 2 days or shorter incubations in a laminar flow hood. Plates may dry unevenly if the laminar air flow is parallel to the bench. Use of a template makes it easier to spot yeast cells at regular distances. Two sample templates are provided in Figure 1. These may be printed, cut out, and affixed to the inside of a Petri dish lid. - Allow plates to dry on the bench top.
- Incubate plates at 30 °C for 2 – 6 days.
- Photograph each plate after incubation.
Figure 1. Templates for spotting yeast cells onto 100-mm agar plates. These templates may be used to facilitate spotting yeast at regular distances with a multichannel pipettor. Templates may be printed, cut out, and affixed to the inside of a Petri dish lid. Place Petri dish with growth medium inside lid with template affixed. Templates are marked with a notch to track orientation. It is recommended that plates used in growth assays be similarly marked with a notch to track orientation. Templates for spotting four (A) or five (B) serial dilutions of yeast cells are provided. Please click here to view a printable version of this figure with 100-mm templates.
2. Biochemical Confirmation of Yeast Growth Assay
- Growth of Yeast Cells and Post-Alkaline Protein Extraction (modified from 31)
- Transform wild-type and mutant yeast cells with a plasmid encoding the unstable protein.
- Inoculate transformants in 5 ml of SD medium that is selective for cells harboring plasmid molecules. Incubate overnight at 30 °C, rotating.
- Measure the OD600 of each overnight culture.
NOTE: Following overnight incubation, cells may be in either logarithmic or stationary growth phase but should have reached an OD600 that will permit dilution to an OD600 of 0.2 in 10 ml fresh selective medium (step 2.1.4). Very slow-growing yeast strains may require incubation times longer than one night, or inoculation of a greater number of cells, as determined empirically. - Dilute yeast cells to an OD600 of 0.2 in 10 ml fresh selective medium.
- Continue to incubate cells at 30 °C, rotating or shaking, until cultures reach an OD600 between 0.8 and 1.2 (i.e. are in mid-logarithmic growth).
NOTE: If the unstable protein of interest is under the control of a regulatable promoter, the optimal timing of induction of protein expression and cell harvest may vary according to previous studies or empirical observations. - Collect 2.5 OD600 units of culture in a 15-ml conical tube by centrifugation at 5,000 x g for 5 min at room temperature. Remove supernatant by pipetting or aspiration.
NOTE: One OD600 unit is defined as the amount of yeast present in 1 ml of culture at OD600 of 1.0. The volume of culture (in ml) required to harvest 2.5 OD600 units (V) can be determined using the following equation: V = 2.5 OD600 units / Measured OD600 - Resuspend cells in 1 ml distilled water. Transfer suspended cells to a microcentrifuge tube.
- Pellet cells by centrifugation at 6,500 x g for 30 sec at room temperature. Remove supernatant by pipetting or aspiration.
- Resuspend cells in 100 µl distilled water by pipetting up and down or vortexing, and add 100 µl 0.2 M NaOH. Mix by pipetting up and down. Incubate samples for 5 min at room temperature.
- Pellet cells (most of which have not yet released proteins and are still viable) by centrifugation at 18,000 x g for 5 min. Remove supernatant by pipetting or aspiration.
- Resuspend pellet in 50 – 100 µl 1x Laemmli sample buffer, which will lyse cells, by pipetting up and down or vortexing.
NOTE: Removal of the alkaline supernatant following centrifugation and subsequent resuspension of cells in Laemmli sample buffer extracts proteins at a pH compatible with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a Tris-glycine running buffer system and western blotting. - To fully denature proteins, incubate lysates at 95 °C for 5 min.
NOTE: Aggregation-prone proteins (e.g. proteins with several transmembrane segments) may become insoluble when incubated at 95 °C. Therefore, lysates should be incubated at lower temperatures (e.g. 37 °C – 70 °C) for 10 – 30 min, as empirically determined, for the analysis of such proteins. - Cool lysates by placing on ice for 5 min.
- Centrifuge lysates at 18,000 x g for 1 min at room temperature to pellet insoluble material. Separate the supernatant (solubilized extracted protein) by SDS-PAGE prior to subsequent western blot analysis (section 2.2). Alternatively, store lysates at -20 °C.
- Representative Western Blotting Protocol
- Load empirically determined volume of lysates in an SDS-PAGE gel.
- Run gel at 200 V until dye front has reached the bottom of the gel.
- Transfer proteins from gel to polyvinylidene fluoride (PVDF) membrane by wet transfer at 20 V for 60 – 90 min at 4 °C.
- Block membrane by incubating in 5% skim milk in Tris-Buffered Saline (TBS), rocking, for 1 hr at room temperature or overnight at 4 °C.
- Decant blocking solution.
- Incubate membrane with primary antibody specific for protein of interest (or epitope tag thereof) in 1% skim milk in TBS with 0.1% Tween-20 (TBS/T) for 1 hr at room temperature, rocking.
- Decant antibody solution, and wash membrane 3 x 5 min with TBS/T at room temperature, rocking.
- Incubate membrane with appropriate fluorophore-conjugated secondary antibody in 1% skim milk in TBS/T for 1 hr at room temperature, rocking.
NOTE: Because fluorophores are light-sensitive, dilutions of fluorophore-conjugated antibodies should be prepared in the dark. Additionally, incubation of membranes in the presence of fluorophore-conjugated antibodies should occur in lightproof containers. This can be accomplished by wrapping incubation trays in aluminum foil. - Decant antibody solution, and wash membrane 3 x 5 min with TBS/T at room temperature, rocking.
- Acquire image of membrane using Li-Cor Odyssey CLx and Image Studio software (or comparable imaging equipment and software), according to manufacturer recommendations.
- After imaging membrane, incubate the membrane with a primary antibody specific for a loading control protein in 1% skim milk in TBS/T for 1 hr at room temperature, rocking.
- Decant antibody solution, and wash membrane 3 x 5 min with TBS/T at room temperature, rocking.
- Incubate membrane with appropriate fluorophore-conjugated secondary antibody in 1% skim milk in TBS/T for 1 hr at room temperature, rocking.
- Decant antibody solution, and wash membrane 3 x 5 min with TBS/T at room temperature, rocking.
- Acquire image of membrane using Li-Cor Odyssey CLx and Image Studio software (or comparable imaging equipment and software), according to manufacturer recommendations.
To illustrate this methodology, the His3 enzyme has been fused to the carboxy-terminus of the model endoplasmic reticulum (ER)-associated degradation (ERAD) substrate, Deg1-Sec62 (Figure 2A) to create Deg1-Sec62-His3 (Figure 3). Deg1-Sec62 represents a founding member of a novel class of ERAD substrates that are targeted following persistent, aberrant association with the translocon, the channel primarily responsible for moving proteins across the ER membrane32-34. Such unstable proteins have provisionally been called ERAD-T (for translocon-associated) substrates. Previous studies indicate that, upon aberrant translocon engagement, Deg1-Sec62 is targeted for degradation by the Hrd1 ubiquitin ligase (Figure 2B-D)32,34,35. Factors required for the degradation of other Hrd1 substrates appear to be dispensable for Deg1-Sec62 degradation, suggesting a novel degradation mechanism32. Under conditions of impaired lipid binding and prolonged translocon association, apolipoprotein B, the protein component of mammalian low-density lipoproteins, appears to be degraded by a related mechanism36-38. Therefore, Deg1-Sec62 may provide a useful model for degradation of medically relevant translocon-associated proteins.
Wild-type and hrd1Δ yeast cells that lack the chromosomal HIS3 gene were transformed with an empty vector39 or a plasmid encoding Deg1-Sec62-His3 and spotted onto selective growth medium (Figure 4). To confirm that equal numbers of transformed yeast cells were transferred to plates, cells were spotted onto medium lacking tryptophan (which selects for cells harboring the plasmid) but containing histidine. Similar growth was observed for all transformed yeast cells. Cells that express Deg1-Sec62-His3 were expected to grow in the absence of histidine only when the fusion protein is stabilized (i.e. when ERAD-T is compromised). Indeed, hrd1Δ yeast expressing Deg1-Sec62-His3 exhibited a growth advantage relative to wild-type cells expressing Deg1-Sec62-His3 on medium lacking tryptophan and histidine. However, marked fusion-protein-dependent growth in the absence of histidine was observed even in the presence of Hrd1. In order to increase the stringency of the assay, medium lacking histidine was supplemented with 3-amino-1H-1,2,3-triazole (3-AT), a competitive inhibitor of the His3 enzyme40. Yeast expressing Hrd1 grew very poorly on medium lacking histidine supplemented with 1 – 2 mM 3-AT; when HRD1 was deleted, cell growth was restored. Inclusion of 3-AT at a concentration of 3 mM dramatically inhibited growth of all cells, regardless of the presence or absence of Hrd1. These results are consistent with Hrd1-dependent substrate degradation.
Next, the steady-state abundance of the Deg1-Sec62-His3 protein in yeast expressing or lacking the Hrd1 enzyme was directly tested. Western blotting analysis indicated a comparable increase in Deg1-Sec62-His3 and Deg1-Sec62 protein in hrd1Δ yeast relative to wild-type cells (Figure 5). This confirms a role for Hrd1 in the regulation of levels of both proteins. Hrd1-dependent degradation of Deg1-Sec62 proceeds after the protein aberrantly engages the ER translocon32. Importantly, Deg1-Sec62-His3 aberrantly engages the translocon in a similar manner (unpublished data), further validating the use of Deg1-Sec62-His3 as a growth-based reporter for degradation of Deg1-Sec62 specifically and translocon-associated proteins generally.
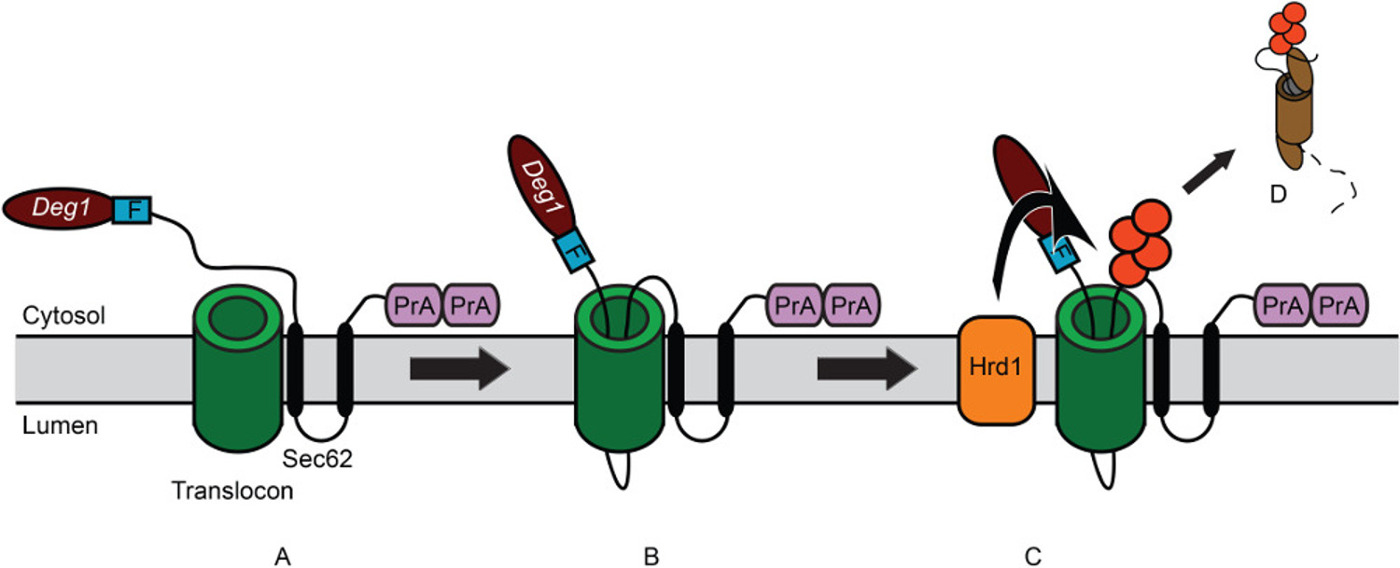
Figure 2: Model for degradation of Deg1-Sec62 following aberrant translocon engagement. (A) Schematic depiction of Deg1-Sec62. Deg1 (the amino-terminal 67 amino acids from MATα2) is followed, in sequence, by the Flag (F) epitope, the 2-transmembrane endoplasmic reticulum (ER) protein Sec62, and two copies of the S. aureus Protein A (PrA). For clarity, the fusion protein is referred to as Deg1-Sec62. (B) Following normal insertion of its two transmembrane segments into the ER membrane, persistent interaction of Deg1-Sec62 with the translocon triggers abnormal, Deg1-dependent translocon engagement. A portion of the initially cytosolic amino-terminal tail aberrantly enters—and likely remains within—the translocon. (C) Following abnormal translocon engagement, Hrd1 recognizes and ubiquitylates Deg1-Sec62. Red circles indicate ubiquitin molecules. (D) Deg1-Sec62 is then extracted from the ER membrane and degraded by the proteasome, likely relieving translocon obstruction.
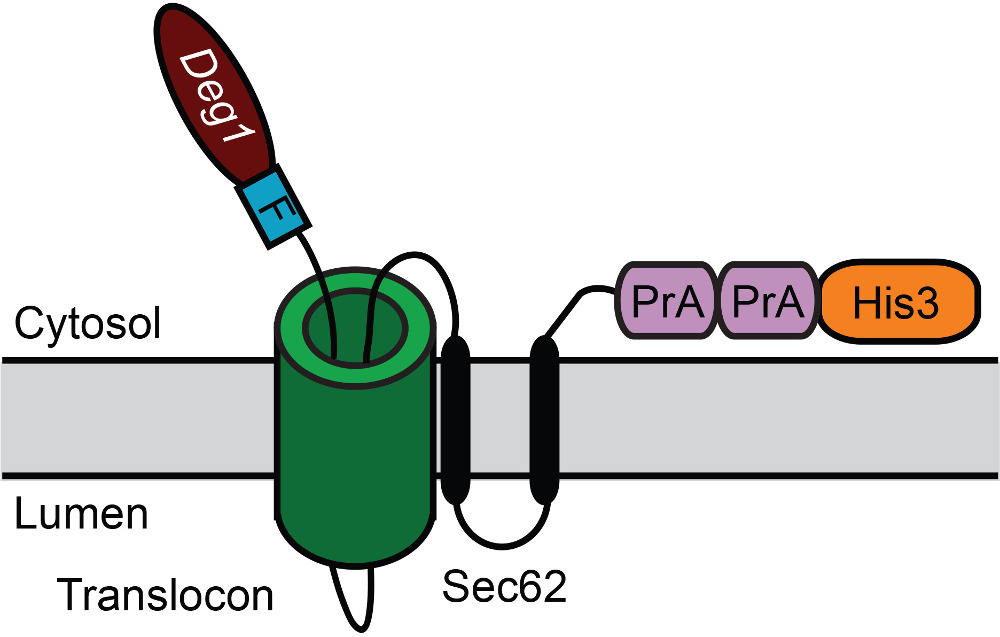
Figure 3: Schematic depiction of Deg1-Sec62-His3 following aberrant translocon engagement. Deg1 is followed, in sequence, by the Flag (F) epitope, the 2-transmembrane ER protein Sec62, two copies of the S. aureus Protein A (PrA), and the yeast His3 enzyme. For clarity, the fusion protein is referred to as Deg1-Sec62-His3.
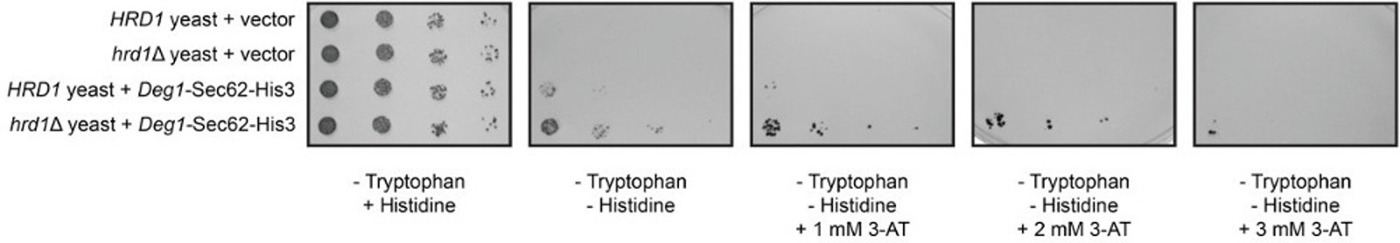
Figure 4: Fusing His3 to Deg1-Sec62 permits selection of degradation-defective mutants. Serial dilutions of wild-type (HRD1) and hrd1Δ yeast transformed with an empty vector or a plasmid encoding Deg1-Sec62-His3 were spotted onto medium lacking tryptophan, medium lacking tryptophan and histidine, and medium lacking tryptophan and histidine supplemented with 3-amino-1H-1,2,3-triazole (3-AT), a competitive inhibitor of His3, at the indicated concentrations. Please click here to view a larger version of this figure.
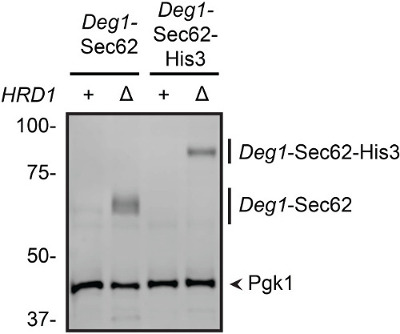
Figure 5: Increased abundance of Deg1-Sec62 and Deg1-Sec62-His3 in cells lacking Hrd1. Protein extracts were prepared from wild-type (+) and hrd1Δ(Δ) yeast expressing Deg1-Sec62 or Deg1-Sec62-His3. Proteins (equivalent to 0.125 OD600 units) were separated by SDS-PAGE, followed by western blotting with rabbit anti-mouse secondary antibodies, which directly bind the Protein A epitopes of the fusion proteins. Subsequent western blotting with antibodies specific to Pgk1 provides a loading control.
| Solution | Components | Comments |
| Synthetic Defined (SD) Minimal Yeast Medium | 2 % dextrose, 0.67 % yeast nitrogen base without amino acids, 0.002 % adenine, 0.004 % uracil, 0.002 % arginine, 0.001 % histidine, 0.006 % isoleucine, 0.006 % leucine, 0.004 % lysine, 0.001 % methionine, 0.006 % phenylalanine, 0.005 % threonine, 0.004 % tryptophan. For solid (plate) medium, include 2 % agar. | 1. Selective medium is prepared by omitting appropriate amino acid(s) or nitrogenous bases. |
| 2. For convenience, these ingredients may be maintained as concentrated stock solutions as follows. Amino acids may be maintained as 100X stock solution containing all desired amino acids. Yeast nitrogen base may be maintained in a 20X stock solution (13.4 %). Dextrose may be maintained in a 40 % stock solution. Adenine and uracil may be maintained as 1 % stock solutions in 0.1 M NaOH. | ||
| 3. Sterilize medium by autoclaving. | ||
| 1X Laemmli Sample Buffer | 2 % SDS, 10 % glycerol, 5 % β-mercaptoethanol, 60 mM Tris HCl pH 6.8, 0.008 % bromophenol blue | 1. 1X Sample buffer is often prepared by diluting a more concentrated (e.g. 5X) stock. |
| 2. The dye bromophenol blue may be added to desired intensity. A "pinch" (very small amount tapped from the edge of a spatula) is typically sufficient. | ||
| 0.2 M Sodium Hydroxide | Prepare in water. Sodium hydroxide reacts with glass. Therefore, for long-term storage, 0.2 M sodium hydroxide should be maintained in plastic containers. | |
| Laemmli Running Buffer (5X) | 125 mM Tris, 960 mM glycine, 0.5 % SDS | To prepare 1 L of 1X Laemmli Running Buffer, dilute 1:5 in dH2O |
| Tris Acetate-SDS Transfer Buffer (5X) | 125 mM Tris acetate (pH 8.8), 960 mM glycine, 0.05 % SDS | To prepare 20 L of 1X Tris Acetate-SDS Transfer Buffer, combine 4 L of 5X stock, 4 L of methanol, and 12 L of dH2O |
| 10X Tris-Buffered Saline (TBS) | 500 mM Tris, 1.5 M NaCl; pH adjusted to 7.5 | To prepare 1 L of 1X TBS, dilute 1:10 in dH2O. 1X TBS may be supplemented with the detergent Tween-20 and powdered skim milk, as appropriate. |
Table 1: Solutions and buffers used in this study.
| Strain Name | Alias | Relevant Genotype | Figures | Source |
| VJY6 | MHY500 | MATa | 4 and 5 | Chen et al., 1993 |
| his3-Δ200 | ||||
| leu2-3,112 | ||||
| ura3-52 | ||||
| lys2-801 | ||||
| trp1-1 | ||||
| gal2 | ||||
| VJY10 | MATa | 4 and 5 | This study | |
| his3-Δ200 | ||||
| leu2-3,112 | ||||
| ura3-52 | ||||
| lys2-801 | ||||
| trp1-1 | ||||
| gal2 | ||||
| hrd1::kanMX4 |
Table 2: Yeast strains used in this study. Details of construction are available upon request.
| Plasmid Number | Full Plasmid Name | Figure | Source |
| pVJ30 | pRS414-PMET25-Deg1-Flag-Sec62-2xProtA | 5 | Rubenstein et al., 2012 |
| pVJ121 | pRS414-PMET25 (empty vector with MET25 promoter) | 4 | Mumberg et al., 1994 |
| pVJ467 | pRS414-PMET25-Deg1-Flag-Sec62-2xProtA-His3 | 5 | This study |
| pVJ477 | pRS414-PGAL4-Deg1-Flag-Sec62-2xProtA-His3 | 4 | This study |
Table 3: Plasmids used in this study. Note that all plasmids contain a yeast centromere to allow replication in yeast cells, the TRP1 gene for selection in yeast cells, and the AmpR gene for maintenance in bacterial cells. Plasmid maps, sequences, and details of construction are available upon request.
