The Importance of Myelin Depletion and Tissue Perfusion
Figure 1 depicts the importance of myelin depletion. Myelin depletion (Steps 5.1 through 5.12 of the protocol above) occurs when the single-cell suspension is incubated in the Myelin Removal Beads, washed, and subsequently passed through the column on the magnetic sorter. The purpose of these steps is to reduce the amount of cellular debris present in each sample. As neurons are dissociated and triturated, it can shear off processes that are typically myelinated, and thus may stain positive with the Thy1 antibody. This results in myelinated debris which can ultimately interfere with the antibody staining and cell sorting. In addition it can reduce the relative number of cells that are retrieved from each sample. In Figure 1, the polygons drawn in each panel of the figure depict the living cells that will be gated and analyzed for subsequent sorting. This gate for living cells is drawn based on the forward scatter (FSC), which is a measure of size, and side scatter (SSC), which is a measure of granularity, of light passing by that particle, which ultimately excludes either dead cells and/or cellular debris. Thus, living cells should have a low level of granularity (low SSC) and a reasonable (not too small or not too large) size (FSC). Sample 1 (Figure 1A) and Sample 2 (Figure 1B) represent two different hippocampal samples that were individually prepared using all steps of the protocol above. The percent of cells that are gated is 53.9% of the total events analyzed in Sample 1 (9,656 cells out of 17,916 events) and 56.1% of the total events analyzed in Sample 2 (9,492 cells out of 16,920 events). Sample 3 (Figure 1C) is also a single hippocampus that was prepared using the protocol above but without the myelin depletion performed in Steps 5.1 – 5.12. The percent of cells that are gated is 3.93% of the total events analyzed in Sample 3 (4,312 cells out of 109,742 events), which is significantly less than in Sample 1 or Sample 2.
Myelin depletion is a relatively new technique, which can be compared in its purpose to a more popular technique, using the density gradient method such as Percoll. The purpose of performing a density gradient method is to remove cellular debris and dying cells from each sample. While the density gradient method is effective to some extent in this purpose (see 4,7 for examples of cells sorted following a density gradient method); it does not work nearly as well as the method of myelin depletion. The data in Figure 1 demonstrate the importance of myelin depletion in the increased total yield of cells. In addition, antibody staining can be greatly enhanced with the removal of myelinated debris.
In this protocol, it is important to perfuse the blood from the neural tissue using cardiac perfusion prior to tissue dissection and collection. This significantly helps to reduce the amount of neural cells that die during the procedure. Typically blood is separated from neural tissue by the blood brain barrier; however, dissection and dissociation of the tissue without perfusion can allow direct contact between neural cells and blood, which can result in increased neural cell death.
Staining Controls
Figure 2 represents the examples of the staining controls that must be performed each time cells are sorted. In this protocol, representative samples were stained with three antibodies including an APC-conjugated CD11b antibody to identify microglia, a FITC-conjugated Thy1 antibody to identify neurons, and an anti-GLT1 primary antibody with a phycoerythrin (PE) -conjugated secondary antibody to identify astrocytes. First, cells are sorted based on their forward and side scatter from all possible events. This gate is identified as P1 (population 1, Figure 2A). Next, the single cells, also called singlets, are sorted based on their size from any doublets or larger clumps of cells (see Figure 2A). These cells are not stained (and thus not gated) with any of the antibodies listed above. Instead, the gates for each cell type are determined based on the staining of the “single stain” controls that are prepared for each antibody. Figure 2B and Figure 2C depict the gate for APC-CD11b positive cells that are not present in the blank (unstained) control (Figure 2B) but are present in the APC alone (single stain) control sample. Figure 2D and Figure 2E depict the gate for the PE-GLT1 positive cells that are present in the GLT1-PE single stain control but not present in the FITC-Thy1 single stain control. Figure 2F depicts the gate for PE-GLT1 positive cells and FITC-Thy1 positive cells that are not present in the blank (unstained) control (similar to Figure 2B, which contains no stained cells in the CD11b positive gate). Figure 2G depicts the lack of both GLT1-PE staining and the lack of FITC-Thy1 staining in the APC alone (single stain) control. Figure 2H represents the PE-GLT1 positive cells that are present in the GLT1-PE single stain control (similar to Figure 2D but with a different axis for comparison), while Figure 2I represents the FITC-Thy1 positive cells found in the FITC-Thy1 single stain control. These single stain and unstained controls are used to identify the positive and negative cell populations for each antibody, to perform compensation in case one fluorochrome interferes with the signal from another fluorochrome, and to confirm that these antibodies work individually before they are combined in the samples themselves.
Sorting Neurons, Astrocytes, and Microglia from Individual Samples
Figure 3 depicts the sorted populations of neural cells from a single male hippocampus obtained from a FACS machine, and Figure 4 depicts the sorted populations of neural cells from a single male cerebellum. As described above, cells are sorted based on their forward and side scatter from all possible events (see Figure 3A and Figure 4A). This gate is identified as P1 (population 1). Next, the single cells, also called singlets, are sorted based on their size from any doublets or larger clumps of cells (see Figure 3B and Figure 4B). This gate is identified as P2. It is necessary to note the importance of obtaining singlets from the cell population. In order to accurately sort individual cell types, it is critical that the cells are not bound to each other in clumps of two or more. This would ultimately prevent the proper sorting of pure populations of cells. In order to prevent the appearance of doublets or larger clumps of cells in the samples, one must be sure to triturate each sample thoroughly and consistently across all samples with the glass Pasteur pipettes (numbered 1, 2, and 3) as described in Steps 4.8 through 4.11. Third, the single cells are then gated as either APC-CD11b positive (CD11b+ gate, P4, see Figure 3C and Figure 4C) or APC-CD11b negative (CD11b- gate, P3, see Figure 3C and Figure 4C). APC-CD11b negative cells are subsequently sorted into PE-GLT1 positive cells (GLT1+ gate, P6) and FITC-Thy1 positive cells (Thy1+ gate, P5), see Figure 3D and Figure 4D. Following each sort, the analysis program will determine the total number of events in each gate or population as well as the percent of the parent population(s). NOTE: We have used the FACS software in the current examples.
Confirming the Purity of each Cell Population using Real-time Polymerase Chain Reaction (PCR) of Cell-type-specific Genes
Figure 5 depicts the real-time PCR results obtained from the male hippocampus sample depicted in Figure 3 plus three additional male hippocampal samples (n = 4 hippocampi) and from the male cerebellum sample depicted in Figure 4 plus three additional male cerebellum samples (n = 4 cerebellum). To confirm the purity of the sorted cells in this particular example, we analyzed the relative gene expression of alternative cell-type-specific genes, using ribosomal 18s as the housekeeping gene for standardization. We analyzed the relative expression of the ionized calcium-binding adapter molecule (Iba1) which is a cytoplasmic protein expressed exclusively in microglia within the brain. We also analyzed the relative expression of the glial fibrillary acidic protein (GFAP) which is an intermediate filament protein expressed in astrocytes, and we also analyzed the relative expression of the N-methyl-D-aspartate (NMDA) receptor subunit 1 (NR1), which is expressed predominantly on neurons. These real-time PCR results confirm the purity of the sorted cells and identify some interesting differences across brain regions. For example, as expected, Iba1 was expressed approximately 400-fold higher in microglia than it was in either neurons or astrocytes (see Figure 5A and Figure 5D). As expected, GFAP was expressed more than 2,000-fold higher in hippocampal astrocytes than it was in hippocampal neurons or microglia (Figure 5B). Interestingly, however, GFAP was not highly expressed in the neurons or even the GLT1+ astrocytes that were sorted from the cerebellum as expected (Figure 5E). Given that others have identified GFAP expression in the cerebellum7; there are two possible explanations for these results. First, it is possible that GLT1 is expressed on a different subtype of astrocytes, one that does not co-express GFAP in the cerebellum or it is possible that GLT1 is not expressed at all on astrocytes in the cerebellum and that a different type of cell, that is neither Thy1+ or CD11b+, has been isolated from the cerebellum using this technique. Finally, as expected, NR1 was expressed approximately 300-fold higher in neurons than it was in either astrocytes or microglia (see Figure 5C and Figure 5F). These representative results confirm and expand upon previous findings using this technique to sort neurons, astrocytes, and microglia with these antibodies from the hippocampus and the nucleus accumbens using a different machine at a different institution1.
Following confirmation of purity of the sort, one can analyze their gene of interest to determine the specific cell type in which it is expressed or whether it is altered following treatment in a cell-type-specific manner. For example, we analyzed the expression of calbindin, a calcium-binding protein that is often used to identify neurons in the hippocampus and the cerebellum. Calbindin was expressed significantly more in hippocampal neurons than in any other hippocampal cell type (Figure 6A) but that it was expressed at significantly higher levels in the neurons isolated from the cerebellum (Figure 6B) than it was from the neurons of the hippocampus.
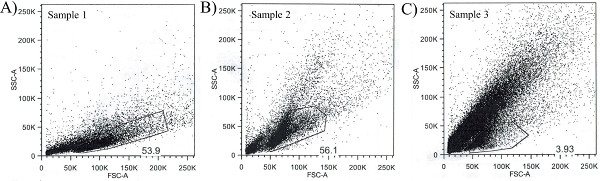
Figure 1. The effect of myelin removal on cell sorting. (A) Sample 1 and (B) Sample 2 represent two different hippocampal samples that were individually prepared using all steps of the protocol above. The percent of cells that are gated is listed inside each example. Specifically, 53.9% of the total events analyzed in Sample 1 were cells (9,656 out of 17,916 events) and 56.1% of the total events analyzed in Sample 2 were cells (9,492 out of 16,920 events). (C) Sample 3 is also a single hippocampus that was prepared using the described protocol, but without the myelin depletion performed in Steps 5.1 – 5.12. The percent of cells that are gated is 3.93% of the total events analyzed in Sample 3 (4,312 out of 109,742 events), which is significantly less than in Sample 1 or Sample 2. Please click here to view a larger version of this figure.
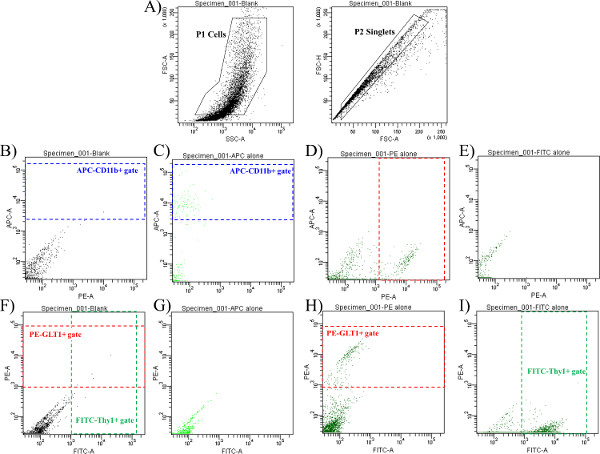
Figure 2. Staining controls must be performed for each sort. Cells were stained with three antibodies including an APC-conjugated CD11b antibody to identify microglia, a FITC-conjugated Thy1 antibody to identify neurons, and an anti-GLT1 primary antibody with a PE-conjugated secondary antibody to identify astrocytes. (A) These plots depict the forward and side scatter of cells that are not stained (and thus not gated) with any of the antibodies listed above. The gates for each cell type are determined based on the staining of the “single stain” controls that are prepared for each antibody below. (B) and (C) depict the gate for APC-CD11b positive cells that are not present in the blank (unstained) control (B) but are present in the APC alone (single stain) control sample (C). (D) and (E) depict the gate for the PE-GLT1 positive cells that are present in the GLT1-PE single stain control (D) but not present in the FITC-Thy1 single stain control (E). (F) depicts the gate for PE-GLT1 positive cells and FITC-Thy1 positive cells that are not present in the blank (unstained) control (similar to (B), which contains no stained cells in the CD11b positive gate). (G) depicts the lack of both GLT1-PE staining and the lack of FITC-Thy1 staining in the APC alone (single stain) control. (H) represents the PE-GLT1 positive cells that are present in the GLT1-PE single stain control (similar to (D) but with a different axis for comparison), while (I) represents the FITC-Thy1 positive cells found in the FITC-Thy1 single stain control. Please click here to view a larger version of this figure.
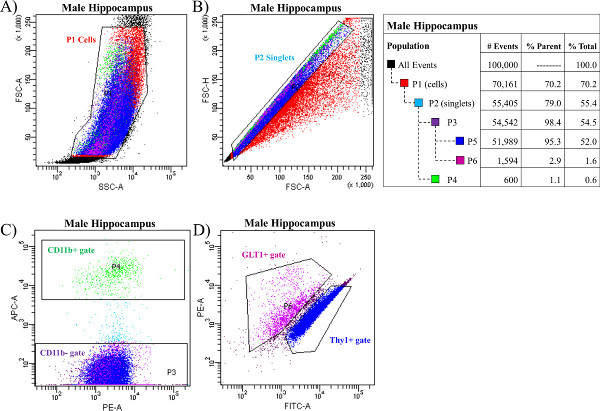
Figure 3. Neurons, astrocytes, and microglia sorted from a male hippocampus. The hippocampus from one male rat was dissociated and stained with the antibodies for CD11b, GLT1 and Thy1 and sorted using a FACS machine. (A) Cells were first sorted based on their forward and side scatter from all possible events. This gate is called P1 (population 1). (B) Next, single cells, also called singlets, were sorted based on their size from the doublets or larger clumps of cells. This gate is called P2. (C) Third, the single cells were gated as either APC-CD11b positive (CD11b+ gate, P4) or APC-CD11b negative (CD11b- gate, P3). (D) APC-CD11b negative cells were subsequently sorted into PE-GLT1 positive cells (GLT1+ gate, P6) and FITC-Thy1 positive cells (Thy1+ gate, P5). The breakdown of all events and all gates was generated from the FACS software depicted in a table which is presented on the right. Please click here to view a larger version of this figure.
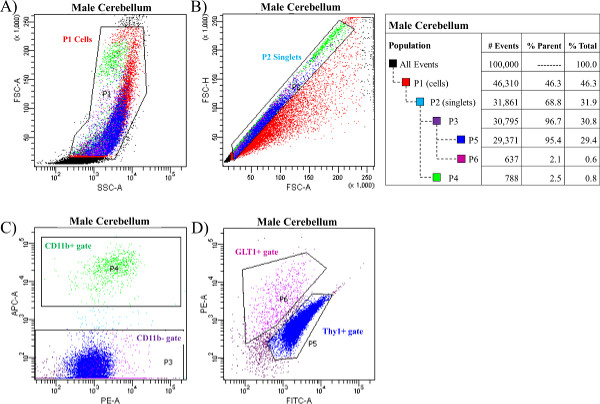
Figure 4. Neurons, astrocytes, and microglia sorted from a male cerebellum. The cerebellum from one male rat was dissociated and stained with the antibodies for CD11b, GLT1 and Thy1 and sorted using a FACS machine. (A) Cells were first sorted based on their forward and side scatter from all possible events. This gate is called P1 (population 1). (B) Next, single cells, also called singlets, were sorted based on their size from the doublets or larger clumps of cells. This gate is called P2. (C) Third, the single cells were gated as either APC-CD11b positive (CD11b+ gate, P4) or APC-CD11b negative (CD11b- gate, P3). (D) APC-CD11b negative cells were subsequently sorted into PE-GLT1 positive cells (GLT1+ gate, P6) and FITC-Thy1 positive cells (Thy1+ gate, P5). The breakdown of all events and all gates was generated from the FACS software depicted in a table which is presented on the right. Please click here to view a larger version of this figure.
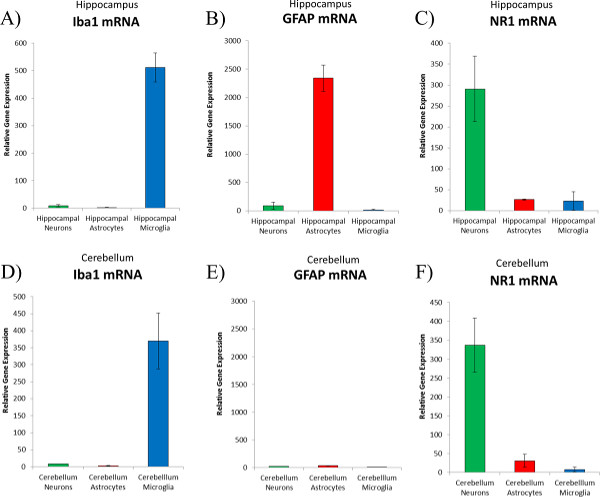
Figure 5. Real-time PCR analysis of cell-type-specific genes from sorted cells. Neurons (green bars), astrocytes (red bars) and microglia (blue bars) were sorted based on the protocol described above and mRNA was extracted for confirmation of cell-type-specific gene expression. (A) Iba1 is a calcium binding protein expressed exclusively in microglia sorted from the male hippocampus. (B) GFAP is a filament protein expressed predominantly in astrocytes sorted from the male hippocampus (C) NR1 is a ubiquitous subunit of the NMDA glutamatergic receptor that was expressed predominantly on neurons sorted from the male hippocampus. (D) Iba1 was also expressed exclusively on microglia sorted from the male cerebellum. (E) Interestingly, GFAP was not expressed in any of the cell types sorted from the male cerebellum. (F) The NR1 subunit of the NMDA receptor was also expressed predominantly on neurons sorted from the male cerebellum. Please click here to view a larger version of this figure.
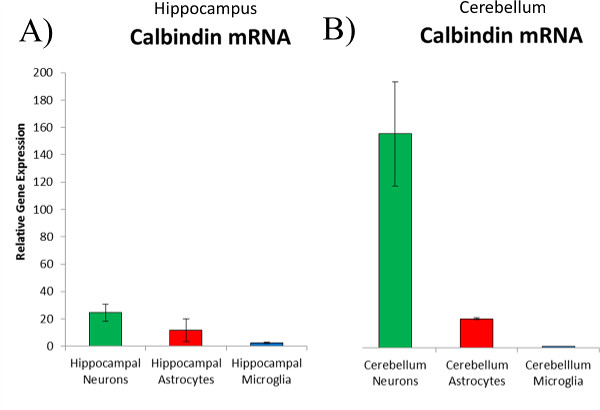
Figure 6. Real-time PCR analysis of calbindin expressed in sorted neural cells. Cells sorted using FACS can be used to analyze cell-type-specific gene expression. (A) Neurons (green bars) expressed significantly more Calbindin than either astrocytes (red bars) or microglia (blue bars) sorted from the male hippocampus. (B) Neurons sorted from the male cerebellum expressed significantly higher levels of Calbindin than either astrocytes or microglia sorted from the cerebellum, but also significantly higher levels than the neurons sorted from the hippocampus. Please click here to view a larger version of this figure.
