Our recent in vivo studies used these live image acquisition and analysis methods to visualize and track the kinetics and patterns of ONH and retinal microglial changes during early stages of chronic glaucoma and their relationship to late neurodegeneration59. Here we illustrate a cSLO image acquisition protocol to visualize microglial cells across a large area of the central retina in individual young heterozygous Cx3CR1-GFP DBA/2J retinas (Figure 5). Based on the high cell resolution, we apply live image thresholding and morphometric analyses that allow the automatic segmentation of individual GFP+ cells and somata (Figure 6).
To monitor the development of local microgliosis and/or microglia activation at ages preceding detectable neurodegeneration in this model of chronic glaucoma, we measured monthly variations of total microglia cell density in individual young heterozygous Cx3CR1-GFP DBA/2J retinas (n = 10) between 3-5 months of age (Figure 7A). Consistent with the variable progression of neurodegeneration in individual eyes41-44, our analysis reveals variable levels of retinal microglia activation and microgliosis across eyes at each age (Figure 7B), and detects dynamic changes in the density of total and activated microglia within individual retinas (Figure 7C). Noticeably, numbers of activated microglia are uncoupled from concurrent changes in total GFP+ cell density. These in vivo findings confirm previous ex vivo analysis of microglial changes in DBA/2J mice45. Thus cSLO live detection and measurement of retinal microglial activation may serve as indicators of early disease progression within individual eyes, and may potentially be linked to initiating events of glaucoma pathogenesis.
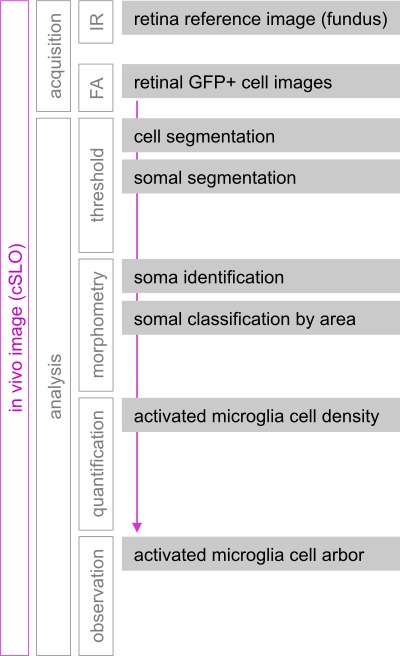
Figure 1: Live image acquisition, processing and analysis workflow. cSLO was used to image hundreds of GFP+ microglial/peripheral monocyte cells localized to the central ~1.7 mm2 of the mouse inner retina. Fluorescence images (FA) were acquired monthly for the same mice, and aligned using infrared (IR) fundus images of the retinal vasculature and vitreal surface as reference. Segmentation of GFP+ cells and somata resulted from applying two separate steps of intensity-thresholding to FA retinal images. Morphometric analysis of the binary representing cell somata allowed measurement of individual somal areas. GFP+ cells with somal area above 50 µm2 were classified as activated. Density of total and activated GFP+ cells was measured for individual retinal FA images, and examination of arbor extension and branching complexity was performed by direct observation of FA images.

Figure 2: Live image acquisition controls in cSLO Spectralis. (A) Manual touch screen panel display on initiation. (B) cSLO software imaging window, showing the head of a live mouse. (C) Settings selected for infrared imaging (lit in blue). (D) Fundus image of the retina during real time acquisition (left panel) and average (right panel). Normalization command indicated with black circle. Arteries and veins radiating from the optic nerve head are visible (between brackets), as are the optic axonal bundles (arrows) in between the blood vessels. (E) Settings selected for fluorescence imaging within a single xy point. (F) Single-point fluorescence (FA, 488 nm) imaging during acquisition (averaging). The smaller image (left) shows an individual scan, the larger image (right) shows the progress of intensity averaging. (G) Settings selected for acquisition of a multipoint (composite), fluorescence image. (H) Multipoint fluorescence imaging acquisition, showing the scan in progress, which the software identifies with a green circle (or red when acquisition is not possible). Scale bars represent ~5 mm (B) or 250 µm (D-H) Please click here to view a larger version of this figure..

Figure 3: Key ocular defects and imaging errors that can prevent correct cSLO imaging of microglial cells. (A–C) Ocular damage or defects, as well as image acquisition errors can prevent reliable imaging of GFP+ cells localized to the innermost planes of the retina. (A) Some gross eye defects are readily detectable in fundus images, including corneal or lens opacity, or injury and deepening of the optic disc area, all of which prevent clear imaging of GFP+ cells. (B) Focusing either above (the walls of the vessels become indistinguishable from the lumen) or below the inner surface of the retina (the vessels are barely visible) reduces the detectability of GFP fluorescence emission from cells located at the nerve fiber and ganglion cell layers. (C) Saturation levels incorrectly or unevenly set can result in bright but oversaturated images (with cells lacking resolution) or images with obscured areas. Scale bar represents 250 µm. Please click here to view a larger version of this figure.

Figure 4: Binary thresholding-based quantification of microglial cell counts and somal area. (A–D) Steps of cell segmentation in FluoRender. (A) Navigation window with rendered view of a representative single-point fluorescence image of microglia within the central retina (top panel); corresponding rendering properties (lower panel). (B) The same image during a cell segmentation routine, viewed as RGB, with corresponding threshold settings (lower panels). Thresholded cells are represented by white pixels and overlaid to their processes in cyan. (C) Image of cell masks obtained after cell segmentation. (D) Previous mask with inverted greyscale values for further image processing. (E–H) Steps of somal thresholding and quantification. (E) Use of intensity threshold to segment microglia somata based on their previous cell segmentation. The RGB intensity histogram (left) allows thresholding by direct selection of the lower 50-60% range of intensities (0 to 127-153). This segmentation creates a quantifiable mask that isolates individual microglial somata (blue). Images are rescaled using the spline function (oval at right). (F) Automatic threshold-based measurements of individual ROI/somata (top panel) and ROI silhouettes (Object Catalog, bottom panel) used to correct overlapping ROIs using Binary Tools (right panel) for manual separation or erosion. (G) Classification of individual somal areas by Area Restriction (top left panel). Segmented somata are classified as ROIs smaller and larger than 50-60 µm2, and the binary layers for each class of somata are assigned different color pixels (green and magenta, respectively). (H) Overlay of the segmented somata (green) on the original image (with increased contrast) used for direct observation of process complexity in individual activated cells (left panel). Magnified view of cell arbors. Scale bars represent 250 µm (A-H) or 50 µm (H, inset). Please click here to view a larger version of this figure.

Figure 5: High-resolution visualization of CX3CR1-GFP+ microglia/monocytes localized to the inner mouse retina by live confocal scanning laser imaging. (A) Single xy-point cSLO image showing GFP+ cells within the central retina in a 2 month old DBA/2J mouse (left). Magnified view (right) showing the ONH area (circle), blood vessels (lines) lined with perivascular microglia/macrophages, and parenchymal microglia tiling the central retina. (B) Multipoint image, collected immediately after the single point to visualize microglia across a wider retinal area (⅓ of the retina). (C, D) Identical images, shown with inverted greyscale for optimal observation of cell morphology and complexity (minimally adjusted for contrast and resolution). Somal size and shape can be resolved in most cells (left). Arbor extension and cell branching can be resolved in the cells with large somata (right, and 3 individual cells). (E) Infrared fundus image of the vasculature and optic disc used for xy-plane alignment of sequential images. Scale bars represent 250 µm (A, B and E), and 25 µm (C, far right). Please click here to view a larger version of this figure.

Figure 6: Segmentation of microglial cell and somata. (A) GFP+ cells rendered by segmentation in 2D using FluoRender. (B) Corresponding cell somata, detected by thresholding using confocal image analysis software. (C) Binary mask of cell somata amenable to quantitative analysis. (D) Classification of segmented cell somata by area to discriminate activated microglial cells (>50-60 µm2, magenta), and non-activated cells (<50 µm2, green). Dim and small cells (<10-20 µm2, yellow asterisk) are identified manually in the original image with inverted greyscale (Figure 3C). Scale bars represent 250 µm. Please click here to view a larger version of this figure.

Figure 7: Longitudinal tracking of microglial cell density and activation in young DBA/2J retinas. (A) Live cSLO fundus and FA images of the same eye, acquired from 3-5 months of age. Sequential FA images were used to quantify changes over time in total numbers of CX3CR1-GFP+ cells localized within the central ~1.7 mm2 of retina. Counts included parenchymal and perivascular microglia, but excluded cells clustered at the ONH (position indicated with white circle). (B) Timecourse analysis of retinal microglia cell density and activation at ages preceding neurodegeneration in DBA/2J chronic glaucoma (n = 10 retinas per age). Numbers of total GFP+ cells and of activated microglial cells (somal area >50 µm2) per mm2 of central retina. Each data point represents a single retina, and means for each age are represented with a horizontal line. (C) Monthly tracking of numbers of total GFP+ cells and of activated microglial cells in a single retina. There are parallel but quantitatively different changes in total and activated cell density, with more dynamic variation in the density of morphologically activated cells. Scale bar represents 250 µm (A). Please click here to view a larger version of this figure.
