1. Preparation of Multi-phenotype Assay Plate (MAP) Substrates, Basal Media, Pre-growth Media, and Buffer
- Make 50 ml of 1.0 – 1.25% substrate stocks.
- Dissolve 1.0 – 1.25% (w/v) of solid substrate in sterile water (heat if necessary). Filter sterilize solutions with a 0.22 µm filter unit and store stocks at RT. Examples of substrates used in these experiments are provided in Table 2.
- Make 250 ml of 3x basal media for all substrate classes (carbon, nitrogen, sulfur and phosphorus). The MOPS (3-morpholinopropane-1-sulfonate) based basal media22 contains the following: 1x MOPS (40 mM MOPS + 10 mM Tricine), 0.4% glycerol*, 9.5 mM NH4Cl*, 0.25 mM NaSO4*, 1.0 mM MgSO4*, 1.32 mM K2HPO4*, 10 mM KCl, 0.5 µM CaCl2, 5 mM NaCl, 6 µM FeCl3, and 0.1 % (w/v) L-arabinose** (Tables 1 and 2).
Note: *These compounds are not included depending on the basal media. For example, 0.4% glycerol is not used in the carbon basal media and in the sulfur basal media 1.0 mM MgSO4 is replaced by 1.0 mM MgCl2. **L-arabinose is specific for induction of pBAD promoter vector, pEMB11, which was transformed into an ara–E. coli K-12 strain (BW 27784)23.- Add each component of the basal media to a sterile flask and bring solution to volume. Mix well. With a 0.22 µm filter unit, filter sterilize each basal media into a sterile bottle.
- Make 500 ml of MOPS pre-growth media. MOPS based pre-growth media22 contains the following: 1x MOPS (40 mM MOPS + 10 mM Tricine), 0.4% glycerol, 9.5 mM NH4Cl, 0.25 mM NaSO4, 1.0 mM MgSO4, 1.32 mM K2HPO4, 10 mM KCl, 0.5 µM CaCl2, 5 mM NaCl, and 6 µM FeCl3.
- Add each component of the pre-growth media to a sterile flask and bring to volume. Filter sterilize with a 0.22 µm filter unit, then add ampicillin at a final concentration of 100 µg/ml. Store solution at 4 °C.
- Make 500 ml of 0.5x Luria-Bertani (LB) agar plates. To a sterile flask, add LB components to make a 0.5x concentration (1.25 g yeast extract, 2.5 g NaCl, 2.5 g tryptone, 7.5 g agar). Mix well and autoclave to sterilize.
- Cool agar, then add 100 µg/ml of antibiotic ampicillin with mixing. Pour ~25 ml of agar into Petri dishes, allow to set, and store at 4 °C until needed.
- Make 250 ml of 10 mM Tris (pH 7.4) plus 10 mM MgSO4 buffer solution. Filter sterilize with a 0.22 µm filter unit, and store solution at RT.
2. Preparation of Bacterial Cell-suspension
- Prepare fresh colonies. From a 12.5% glycerol frozen stock, plate fresh E. coli clones onto 0.5x LB agar containing 100 µg/ml ampicillin. Incubate at 37 °C for 24 hr.
- Prepare liquid cultures:
- Pipette 3 ml of pre-growth media containing 100 µg/ml ampicillin into culture tubes. For each clone, use biological triplicates.
- Inoculate independent colonies from section 2.1 into prepared culture tubes. Incubate at 37 °C with shaking for 22 hr.
- Pellet and wash bacterial cells:
- Transfer O/N cultures into 1.7 ml microfuge tubes. Pellet cells via centrifugation at maximum speed (16,900 x g) for 2 min in a microcentrifuge. Decant supernatant and wash cells by re-suspending in 500 µl of 10 mM Tris/10 mM MgSO4 buffer. Repeat.
- Re-pellet cells, decant supernatant and re-suspend cells in 1 ml of 10 mM Tris/10 mM MgSO4 buffer.
- Measure cell density and concentrate cells (if necessary):
- Dilute cells from section 2.3.3 10-fold in the 10 mM Tris/10 mM MgSO4 buffer and transfer this dilution into a cuvette. Measure optical density (OD600 nm) of this dilution and record.
- Concentrate cells to achieve final concentration of 0.07 (OD600 nm) in the MAPs (the cells will be diluted 15 fold when they are added to the MAPs, therefore cells need to be at an initial concentration of OD600 nm = 1.05). Transfer concentrated cell suspension into a reservoir and save (at RT) until step 3.5 of MAPs preparation.
3. Preparation of Multi-phenotype Assay Plates (MAPs)
- Label MAPs. Label sterile 96-well micro-titer plates with an E. coli clone identification number and MAP schematic type.
- Aliquot sterile water into MAPs. Aseptically transfer sterile water into a liquid reservoir. Pipette 60 µl into each well of the micro-titer plate using a multi-channel pipette.
- Aliquot basal media into MAPs. Aseptically transfer 3x basal media into a liquid reservoir. Pipette 50 µl into each well of the micro-titer plate using a multi-channel pipette. Repeat steps 3.2 and 3.3 for each basal media used in the MAP schematic.
- Aliquot substrates into MAPs. Transfer 30 µl of each substrate into the appropriate well of the MAPs.
- Add bacterial suspension to the MAPs. Transfer 10 µl of bacterial cells from section 2.4.2 into each well of the MAP using a multi-channel pipette. Change tips before re-introducing pipette back into the culture stock.
- Cover MAPs with adhesive film. Cover each plate with a single adhesive plate film. Firmly press the film atop the wells of the plate and along the edges to create an even and tight seal. Remove excess film from the edges of the micro-titer plate with a sterile razor blade.
- Measure optical density of MAPs:
- Place prepared MAPs in the “Input” sleeve of a multi-plate spectrophotometer plate reader. Open plate reader software and create a protocol that measures absorbance (OD600 nm) every 30 min, for a total of 32 hr, with 60 sec of shaking between reads.
- Set temperature in the apparatus to 37 °C. Start protocol once MAPs have equilibrated to set temperature.
- After 32 hr, remove MAPs from the “Output” sleeve of the plate reader. Label each data text file to include: the E. coli clone identification number, date the MAP ran, and the MAP schematic type.
4. Multi-phenotype Assay Plates (MAPs) Processing and Parameterization
- Assess growth curves using an automated QA process, e.g., PMAnalyzer24.
- Verify that growth curves have OD600 nm < 0.20 in the first 2 hr. Do not use absorbance at the initial time point (t0) in the filter due to artifacts of condensation, which typically resolve at t1 (30 min).
- Remove growth curves that violate QA filter. Save curve information (sample name, well number, and OD600 nm values) in a separate output file for future reference. Continue with analysis if growth curve passes QA filter.
- Calculate median growth curves. Using replicate data, per well, calculate the median growth curve by taking the median optical density (OD600 nm) value at each time point. Record resulting median growth curves in a separate output file for future use.
- Calculate the best-fit logistic model for each growth curve using an adaptation of the equation proposed by Zwietering25. The logistic equation includes three parameters describing bacterial growth: the lag time, λ (hr), the maximum rate of growth, µmax (OD600 nm 100 -1), and the final biomass yield, α (OD600 nm). Use data at time = 30 min (y1) as the initial value due to artifacts of condensation.
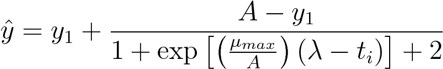 [1]
[1]
- Calculate the maximum growth rate using a direct search. Record the largest rate of change over a 90 min interval within the data.
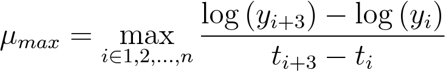 [2]
[2] - Estimate the final biomass yield by searching for the upper asymptotic value of the largest moving average over a 90 min window. Specifically, define as the asymptote of the growth curve.
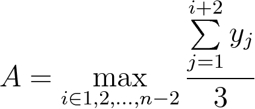 [3]
[3] - Using the µmax and A values from 4.3.1 and 4.3.2, find the λ value by trying all values λ:
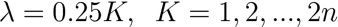 [4]
[4]
- Use each λ value to calculate a sum of squared error (SSE). Use the lowest SSE is the SSE (λS):
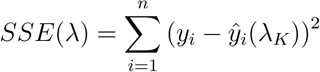 [5]
[5]
- Use each λ value to calculate a sum of squared error (SSE). Use the lowest SSE is the SSE (λS):
- Calculate the maximum growth rate using a direct search. Record the largest rate of change over a 90 min interval within the data.
5. Phenotype Analysis of MAPs
- Calculate the growth level (GL) for each growth curve to assess the overall growth per clone per substrate. Define GL as the harmonic mean of the shifted logistic-fitted values:
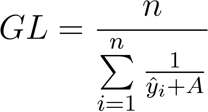 [6]
[6] - Assign a phenotype to each growth curve based on the GL values. Here, the four phenotypes used are: expected growth, no growth, gain of function, and loss of function.
- Determine phenotypes by comparing growth across all clones on a specific substrate in terms of standard deviations away from the mean. Use this statistic and a minimum growth threshold to designate the phenotype presented by the growth curve (Figure 1A,B). Figure 1C provides examples of phenotype assignment.
- Define growth as GL ≥ 0.4 (Figure 1A,B). Define Gain of Function (Figure 1C, green) as having a GL > two standard deviations above the mean and a mean > the growth threshold. Loss of Function (Figure 1C, red) is defined as having a GL < two standard deviations below the mean and a mean > the growth threshold.
- Create visual representations of the data set based on phenotypes and growth curves.
- View growth dynamics portraying OD600 nm values as color to quickly compare growth curves between multiple clones (Figure 2).
- View phenotypic distributions among all clones based on growth conditions (Figure 3).
6. Building the Continuous Culture Apparatus
- Reactor port construction (Figure 4A,B). Drill three 1/4 in. holes, spaced 3/4 in. apart, in the cap of the continuous culture reactor.
- For port α: screw a female luer thread style panel mount to 1/16 in. barb from the bottom of the cap with the barb pointing up, out of the cap. Secure barb with a 1/4 in. panel mount lock nut.
- For port β: screw a female luer thread style panel mount to 1/16 in. barb with the barb pointing up, out of the cap. Secure barb with a 1/4 in. panel mount lock nut.
- For port γ: screw a female luer thread style panel mount to 1/16 in. barb from the bottom of the cap so the barb is pointing up, out of the cap. Secure barb with 1/4 in. panel mount lock nut. Screw a male luer with integral lock ring to 1/8 in. wide bore hose to the barb fitting on the inside of the cap.
- For the outflow port: drill a 5/16 in. hole at the 75 ml mark of the continuous culture reactor. Cut 3/4 in. off the nut end of the 1/4 in. barbed bulkhead fitting. Screw in the 1/4 in. barbed bulkhead fitting (gasket is on the outside of the bottle and the nut is on the inside, Figure 4B).
- Feeding bottle port construction (Figure 4C). Drill two 1/4 in. holes spaced 1 in. apart in the cap of the feeding bottle.
- For the port δ: screw a female luer thread style panel mount to 1/8 in. barb with the barb pointing up, out of the cap. Secure barb with a 1/4 in. panel mount lock nut.
- For the port ε: screw a female luer thread style panel mount to 1/16 in. barb with the barb pointing up, out of the cap. Secure barb with a 1/4 in. panel mount lock nut.
- Screw a male luer with integral lock ring to 1/8 in. wide bore hose to the barb fitting, on the inside of the cap.
- Feeding tubes and tube extensions:
- Cut two 1 in. pieces of tubing (1/8 in. outer diameter (OD) x 1/16 in. inner diameter (ID)). Fit pieces onto ports α and γ of the continuous culture reactor made in section 5.2.
- Cut a single 1 in. piece of tubing (1/4 in. OD x 1/8 in. ID). Fit piece onto port Β of the continuous culture reactor.
- Cut a single 3.5 in. piece of tubing (1/8 in. OD x 1/16 in. ID). Fit this piece to the inside of port γ, on the male luer with integral lock ring to 1/8 in. wide bore hose. Port γ is the sampling port of the continuous culture reactor.
- Cut a single 1 in. piece of tubing (1/4 in. OD x 1/8 in. ID). Fit this piece on the δ port of the feeding bottle.
- Cut a single 11 in. piece of tubing (1/16 in. OD x 1/8 in. ID). Fit this piece to the inside of port ε of the feeding bottle, on the male luer with integral lock ring to 1/8 in. wide bore hose.
- Cut two 18 in. pieces of tubing (1/8 in. OD x 1/16 in. ID). Fit one piece to port ε of the feeding bottle. Save the other piece for section 7.
7. Running Continuous Cultures for Metabolomics
- Sterilize materials:
- Autoclave the dry materials for 30 min. Make sure to wrap cap of the feeding bottle made in section 5 in aluminum foil. Keep attached tubing straight.
- Wrap cap of the continuous culture reactor, made in section 5, in aluminum foil. Keep attached tubing straight. Wrap 18 in. tubing cut in section 6.3.5 and the smallest peristaltic pump tubing adaptor in aluminum foil. Keep tubing straight. Autoclave for 30 min.
- Make 2 L of 0.5x LB broth. In the feeding bottle, make 0.5x LB broth. Cover feeding bottle with foil. Autoclave for 1 hr.
- Make 70 ml of 0.5x LB broth. Pour broth into the continuous culture reactor. Place a stir bar in the continuous culture reactor. Cover reactor with foil and autoclave for 30 min.
- Autoclave the dry materials for 30 min. Make sure to wrap cap of the feeding bottle made in section 5 in aluminum foil. Keep attached tubing straight.
- Continuous culture set-up:
- Bacterial cell-suspension.
- From a 12.5% glycerol frozen stock, plate fresh E. coli clones onto 0.5x LB agar containing 100 µg/ml ampicillin. Incubate at 37 °C for 24 hr.
- Inoculate a single colony into 3 ml of 0.5x LB broth containing 100 µg/ml ampicillin. For each clone use biological triplicates. Incubate at 37 °C with shaking for 22 hr.
- Connect the parts together.
- Place all autoclaved materials into a biological safety cabinet and turn ultraviolet light on while media cools. Once media has cooled, add 0.1% L-arabinose and 100 µg/ml ampicillin to all 0.5x LB broth.
- Unwrap continuous culture reactor cap and screw onto the reactor. Avoid touching the sampling tube. Unwrap the feeding bottle cap and screw onto the feeding bottle. Avoid touching the sampling tube.
- Keep the 18 in. tubing attached to port ε sterile. Un-wrap the 18 in. tubing and peristaltic pump tubing adaptor.
- Fit a free end of the 18 in. tubing onto one end of the peristaltic pump tubing adaptor. Fit the other end of the peristaltic pump tubing adaptor to the 18 in. tubing attached to port ε of the feeding bottle cap.
- Screw 0.22 µm filter units onto ports β and δ of the continuous culture reactor. Screw a 0.22 µm filter unit on the adaptor of port α. To the 0.22 µm filter unit, fit the free end of the 18 in. tubing.
- Start the continuous culture.
- In the biosafety hood, inoculate the continuous culture reactor with 100 µl of O/N culture made in section 7.2.1.1.
- Close system before moving the continuous culture into a 37 °C incubator. In the incubator have a magnetic stir plate and mini peristaltic pump set up.
- Fit the peristaltic pump tubing adaptor into the peristaltic pump. Tubing from the feeding bottle starts at the “In” side and tubing leading to the reactor starts at the “Out” side.
- Set the peristaltic pump to: “FAST”. Start the peristaltic pump by switching to “FORWARD”. Check that media begins to flow through the tubing.
- Place reactor on magnetic stir plate and begin mixing. Continuous culture reactor must equilibrate for 24 hr before sampling.
- Sampling of the continuous culture. Screw a 5 ml luer lok syringe onto port γ and pull up 4.5 ml of culture.
- Use 500 µl of the culture to measure the density (OD600 nm). Dilute cells to make 4 x 1 ml cultures at OD600 nm = 0.35.
- Aliquot diluted cultures into 1.7 ml microfuge tubes. Repeat sampling every 12 hr for 4 days.
- Prepare samples for GC-TOFMS:
- Pellet cells via centrifugation at max speed (16,900 x g) for 2 min. Decant supernatant. Wash cells with 500 µl of phosphate buffered saline (PBS). Repeat this step.
- Decant supernatant a final time, and then submerge microfuge tubes with pelleted cells in liquid nitrogen until bubbling stops. Store samples in -80 °C freezer.
- Bacterial cell-suspension.
8. Running Serial Passage Batch Cultures for Metabolomics
- Preparing bacterial cell-suspension. From a 12.5% glycerol frozen stock, plate fresh E. coli clones onto 0.5x LB agar containing 100 µg/ml ampicillin. Incubate at 37 °C for 24 hr.
- Inoculate individual colonies into 3 ml of 0.5x LB broth containing 100 µg/ml ampicillin. Use biological triplicates for each clone.
- Serial passage batch cultures.
- Subculture O/N from 8.1.1 via a 500-fold dilution into 3 ml of LB broth containing 50 µg/ml ampicillin and 0.1% L-arabinose. Subculture should have an OD600 nm < 0.005. Incubate at 37 °C with shaking.
- Check optical density (OD600 nm) at 2 and 2.5 hr. Grow cells to achieve OD600 nm = 0.35.
- Subculture via a 500-fold dilution into 3 ml of LB broth containing 50 µg/ml ampicillin and 0.1% L-arabinose. Subculture should have an OD600 nm < 0.005.
- Check optical density (OD600 nm) at 2 and 2.5 hr. Grow cells to achieve OD600 nm = 0.35.
- Sampling serial passage batch cultures.
- Using sterile forceps, place a 0.22 µm membrane filter on top of a filter manifold. Aliquot 1 ml of Phosphate Buffered Saline (PBS) onto the filter paper and then turn on vacuum pump.
- Repeat addition of 1 ml of PBS. Transfer 1 ml of subculture from section 8.2.3 onto the filter paper. From here, move quickly to get samples into liquid nitrogen immediately. Complete this in 1 min.
- Aliquot 1 ml of PBS onto filter paper to wash sample. Flush rapidly without spilling sample over sides of filter paper. Repeat.
- Using sterile forceps place filter into a 1.7 ml microfuge tube by carefully folding the filter. Submerge microfuge tube in liquid nitrogen until bubbling stops. Store sample in -80 °C freezer.
9. Metabolite Analysis & Processing
- Ship 3 samples per clone on dry ice to a metabolomics core facility for sample processing, analysis and normalization of GC-TOFMS.
- For each sample determine the mean, median, standard deviation and coefficient of variation across metabolites, using replicate data.
- For statistical analysis, manually code a QA pipeline using conditional statements and repetitive executions26 to remove metabolites that do not follow defined conditions.
- For Condition 1, remove metabolites in which more than 2 samples have zero abundance. Remove internal standards from the metabolomic profiles.
- For Condition 2, remove those metabolites that are not found in the cells of interest. Here, remove the following metabolites: indole 3 acetate, dihydroabietic acid, nonadecanoic acid, salicylic acid, salicylaldehyde, cholesterol, phenol, 1-monostearin, octadecanol, 1-monopalmitin, dodecane, dodecanol, 1-hexadecanol, 5-methoxytryptamine, benzoic acid, pentadecanoic acid, phosphoric acid, pelargonic acid, palmitic acid, capric acid, hydroxylamine, stearic acid, myristic acid, maleimide, levoglucosan, and 4-hydroxybutyric acid.
- For Condition 3, remove those metabolites whose coefficient of variation is greater than 1.
- Capture global metabolomics effects by looking at metabolite relative abundances.
- Create a visual representation of the median metabolite abundance per clone for each metabolite (Figure 6).
- Identify outliers by observing clone-metabolite pair frequencies.
- Calculate the Z score for each median value of every metabolite in a clone. Calculate z scores using the following equation:
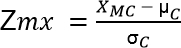 [7]
[7]
Note: Term XMC represents the abundance level of a specific metabolite for a specific clone. Term µC represents the mean of all clones for a specific metabolite. Term σC is the associated standard deviation. - Create a visual representation of the data in which outliers are highlighted (data not provided).
- Calculate the Z score for each median value of every metabolite in a clone. Calculate z scores using the following equation:
All samples used to determine the open reading frames (ORFs) selected in this study were collected from Starbuck Island, Site 7 (STAR7) and Caroline Atoll, Site 9 (CAR9) of the southern Line Islands. An estimated 100 L of seawater from these sites was collected below the coral boundary layer using bilge pumps, as described previously5. Contents of the pumps were subject to fractionalization through large pored filters to remove small eukaryotes and subsequently concentrated using 100 kDa tangential flow filters leaving only microbes and viral like particles (VLPs). To separate the VLPs, remaining seawater was passed through 0.45 µm filters resulting in the virome. Chloroform was introduced to this viral fraction to arrest growth of any remaining cells and stored at 4 °C.
VLPs were purified using the cesium chloride method, in which density gradients separate through centrifugation and allow for the recovery of virions at ~1.35 g/ml to 1.5 g/ml 3. Viral DNA was extracted using a CTAB/phenol:chloroform protocol and amplified via multiple displacement amplification using Phi29 reagents. Sequencing of the virome was accomplished with commercially available pyrosequencing technology.
Bioinformatics used in the processing and selection of viral ORFs for this study are as follows. Three pre-processing steps were used on the CAR9 and STAR7 viral metagenomes. First, public software was used to remove tag sequences that resulted from amplification of the viral DNA prior to sequencing27. Second, common sequencing artifacts such as sequence duplicates and low copy number were filtered out of the dataset via an additional bioinformatics program28. Lastly, removal of foreign sequence contamination29 was performed for those sequences that had ≥ 90% coverage and ≥ 94% identity with sequences in the following databases: RefSeq virus genomes; Human — Reference GRCh37; Human — Celera Genomics; Human — Craig Venter (HuRef); Human — Seong-Jin Kim (Korean); Human — Chromosome 7 version 2 (TCAG); and Human — James Watson, YanHuang (YH; Asian), Yoruba (NA18507; African) reference sequences21. Following these processes, sequences from the CAR9 samples totaled 591,600 and STAR7 sequences totaled 939,311. These sequences were uploaded onto MGRAST and assembled via assembler software using default settings. Contigs were translated into 6 reading frames and putative open reading frames (pORFs) were identified using scripts, as previously described21.
To identify unknown ORFs a number of similarity based searches were performed to remove ORFs of known function. Briefly, the following searches were performed with their corresponding searching criteria21:
- Significant similarity of ≥ 95% identity over ≥ 40 base pair (bp) by MGRAST BLAT in M5NR database.
- Significant similarity (e-value ≤ 0.001) by TBLASTN against all public metagenomes in My Metagenome Database Resource.
- Significant similarity (e-value ≤ 0.001) by BLASTP and TBLASTN against NR database.
- Significant similarity (e-value ≤ 0.001) by RPS-BLAST against the Conserved Domain Database.
- Protein translations from a select fraction of each dataset compared against solved protein structures in the Protein Data Bank.
- Calculation of dinucleotide frequencies using Dinucleotide Signatures package.
The resulting pORFs were designed for expression in E. coli using publically available gene design software. Back-translation of the amino-acid sequences employed a Universal Codon Usage Table designed to accommodate expression in E. coli with a minimum usage threshold of 2%. Restriction enzyme recognition sequences for BamHI and HindIII were excluded from the sequences to facilitate cloning. An outside company synthesized the engineered gene sequences30 and then ORFs were cloned into a medium-copy number pBAD promoter vector, pEMB11, via standard restriction enzyme cloning. All clones were transformed into E. coli K-12 strain BW 2778423.
Multi-phenotype Assay Plates (MAPs)
A high throughput and robust software pipeline was leveraged for analysis of the MAPs, the PMAnalyzer24. The pipeline was developed in a Linux server environment and performs various steps including: parsing of optical density files, formatting data into readable text files, pre-processing of growth curves for quality assurance (QA), and performing mathematical modeling techniques to analyze growth curves. The primary modeling scripts were developed in Python version 2.7.5 to make use of the PyLab module.
MAPs reproducibility was assessed using the standard error (SE) for replicate data (Figure 2A). Raw growth curves were compared to logistic growth curves to determine whether the program PMAnalyzer accurately parameterized and modeled clone growth during experimentation (data not shown). For further information on the accuracy and validity of the MAPs and PMAnalyzer see Cuevas et al.24
Following validation of the method, MAPs data was analyzed using the multiple parameters, such as maximum growth rate (µmax) and growth level (GL), provided by the processing pipeline. Comparative visualization of growth curves is often used for growth data interpretation; however, the number of curves that can be visualized at a time for the comparison has limitations. To analyze numerous growth curves simultaneously, heat map derived plots were implored to compare tens of clones grown on a single substrate against that conditions’ average response (Figure 2B). The influence placed by overexpression of a novel phage protein is observed through changes in curve parameters, specifically: lag phase, exponential phase and the maximum biomass yield (asymptote). As an example, the steep climb from lag phase into exponential phase modeled in the growth curve for the Capsid protein (Figure 2A) is reproduced by a quick change in color intensity from black to white for the same clone in the dynamic plot of Figure 2B.
To obtain a global picture of clone distribution across substrates, phenotypic classifications derived from the GL were used (Figure 3). Here, the four phenotypes are separate into four charts where the height of each bar represents the number of clones displaying that phenotype for a specific substrate. Outliers in the data are recognized as clones falling into the “gain of function” or “loss of function” category. Outliers can then be sought out individually and investigated more closely experimentally. In addition, global analysis recognizes substrate biases in the assay. Substrates such as phenylalanine, malic acid, and glycine resulted in a “no growth” classification. Substrates that consistently fall into the no growth classification, across all clones, are not heavily weighted in downstream functional characterization.
Metabolomics
The catabolic products from clones expressing unknown phage genes were identified using metabolomics. Briefly, clones were grown under either a continuous culture or serial passage in batch culture environment prior to being sent out for GC-TOFMS analysis at a metabolomics core facility. For details on sample processing, analysis, and normalization for GC-TOFMS implemented by the chosen core facility see Fiehn et al.31 Briefly, 1 ml of cold extraction solvent is added to each sample, after which samples are vortexed and sonicated in a cold bath for 5 min. Samples are finally centrifuged and half of the sample is decanted and dried down for analysis. Extracts are purified and spiked with internal retention index markers before being loaded onto the gas chromatograph and then subsequently transferred to the mass spectrometer. Data from each sample is analyzed such that signal intensities for all detected signals in the chromatogram are reported. For normalization, the abundance of peaks for each sample is summed and the total peak abundances are averaged between all samples in the set. Metabolite abundances per sample are divided by the sample’s peak abundance and then multiplied by the average peak abundance of the sample set. The resulting data is used for the analysis of metabolomics in the research discussed.
Validation of metabolomics reproducibility was required to determine the appropriate sample size for each culturing method. To detect the precision seen within samples and the variation seen across sample sizes the standard error of the mean (SM) for both n = 3 and n = 6 datasets were reviewed (Figure 5B). Regardless of the continuous culture (CC) sample size, less than 1% of the data had a SM ≤ 1.5. Median SMs were 221 and 300, and values ranged from 0 to 7.55 x 105 and 3.74 x 105, for n = 3 and n = 6 respectively. SMs were also calculated for each set of sample replicates in the serial culture (SC) method. Again, less than 1% of the data had a SM ≤ 1.5, a median SM of 137, and a range from 0 to 3.51 x 105. To compare the SM distributions between each sample set (CC n = 3 vs. CC n = 6, CC n = 3 vs. SC n = 3, and CC n = 6 vs. SC n = 3) a permutation test was performed. The distribution of either continuous culture SM values dataset was not significantly different from the distribution of the serial culture SM values (p-value = 0.0). However, the distribution of SM values for continuous culture n = 3 data was significantly different from that of the SM values for the continuous culture n = 6 data (p-value = 1.908804 x 10-49). Lastly, the coefficient of variation per metabolite was compared before and after implementation of a quality assurance (QA) step (Figure 5C). In total, 210 metabolites were removed after implementation of the QA pipeline (40% of the data). Less than 1% of the removed data had a metabolite abundance of zero, ~2% was internal standard data, ~5% was data from metabolites never before observed in E. coli, and the remaining metabolites (> 30%) had a coefficient of variation greater than 1.
As with the MAPs analysis, global observations provided an initial understanding of the depth of information metabolomics offers. To obtain a global picture, clones were hierarchically clustered based on their relative metabolite abundances providing information on clone-metabolite profiles, potential clones with related functions, and clone-metabolite outliers (Figure 6). To highlight protein functions, metabolites are separated and grouped based on common metabolic pathways. Using this analysis with preliminary results, it was evident that metabolomics is capable of separating genes from different classes (Figure 6, highlighted clones). In addition, outlier identification with the metabolomics data was determined by calculating standard scores (z scores) for each clone-metabolite pair. To ensure statistical significance, outliers were defined as a clone-metabolite pair with a Z score value of 2, accounting for only 5 percent of the data (data not shown).

Figure 1. Definitions of phenotype classifications. (A) Relationship between the growth level (GL) and maximum growth rate. Data points circled in red represent growth curves showing little to no substrate utilization. (B) Boxplot representation defining growth threshold based on the distribution of growth curves with a minimal growth rate (< 0.15 OD/hr). (C) The variance and standard deviation of GL is calculated for substrate D-galactose. Short dashed lines represent two standard deviations away from the mean. Please click here to view a larger version of this figure.

Figure 2. MAPs validation via precision and differentiation. (A) Growth curves for annotated structural (Capsid) and metabolic clones (Thioredoxin), two novel metabolic clones (EDT2440, EDT2441), and the average response of clones grown on sucrose, D-galactose and D-mannose in the MAPs. Blue lines indicate the standard error seen between replicate data (n = 3). (B) The growth curves for 47 different clones are represented as heat maps for sucrose, D-galactose and D-mannose. The annotated structural (green circle) and metabolic clones (orange circle), two novel metabolic clones (dark and light blue circles), and the average response (red circle) are highlighted. Please click here to view a larger version of this figure.

Figure 3. Clone distribution for each phenotype across multiple substrates. The phenotype-clone count for 47 clones across 72 carbon-specific growth conditions. Table provides direct counts for each phenotype. Please click here to view a larger version of this figure.

Figure 4. Diagram detailing the construction of the continuous culture apparatus. (A) Steps used to build ports α-γ of the continuous culture reactor, (B) steps used to build the out flow port of the continuous culture reactor, and (C) the steps to build ports δ and ε of the continuous culture feeding bottle. Please click here to view a larger version of this figure.

Figure 5. Comparison of the phenomic methods presented. (A) Workflow for the preparation of the Multi-phenotype Assay Plates (MAPs), continuous cultures and serial cultures. (B) The percentage of Standard error of mean (SM) counts for both n = 3 and n = 6 sample sizes for the continuous culture (CC) and serial culture (SC) preparation methods for metabolomics. The y-axis is on a log scale. (C) The distributions of coefficients of variation (CV) per metabolite, before and after implementation of the QA pipeline for the continuous culture (CC) method. Please click here to view a larger version of this figure.

Figure 6. Metabolomic profiles of clones grown in continuous culture. The median metabolite abundances for a set of metabolites are plotted for 84 clones grown in continuous cultures. Metabolite profiles for annotated structural (Capsid) and metabolic clones (Thioredoxin), two novel metabolic clones (EDT2440, EDT2441), and the average metabolic response are highlighted in red. Please click here to view a larger version of this figure.
| Compound | Carbon | Nitrogen | Sulfur | Phosphorus |
| Glycerol | − | 0.40% | 0.40% | 0.40% |
| Ammonium chloride | 9.5 mM | − | 9.5 mM | 9.5 mM |
| Sodium sulfate | 0.250 mM | 0.250 mM | − | 0.250 mM |
| Magnesium sulfate | 1.0 mM | 1.0 mM | − | 1.0 mM |
| Potassium phosphate | 1.32 mM | 1.32 mM | 1.32 mM | − |
| Magnesium chloride | − | − | * | − |
| Potassium chloride | 10 mM | 10 mM | 10 mM | 10 mM |
| Calcium chloride | 0.5 µM | 0.5 µM | 0.5 µM | 0.5 µM |
| Sodium chloride | 5 mM | 5 mM | 5 mM | 5 mM |
| Ferric chloride | 6 µM | 6 µM | 6 µM | 6 µM |
| L- arabinose | 0.10% | 0.10% | 0.10% | 0.10% |
| MOPS pH 7.4 | 1x | 1x | 1x | 1x |
Table 1. The compounds and concentrations of the different basal media used in the MAPs. *1.0 mM of magnesium chloride is substituted. 1x MOPS = 40 mM MOPS, 4 mM Tricine.
| Carbon substrates | Nitrogen substrates | Sulfur substrates | Phosphorus substrates |
| 2 deoxy-D-ribose | 2-deoxy-D-ribose | 1-butane-sulfonic acid | adenosine-5-monophoshate |
| 4 hydroxy-phenyl acetic acid | acetamide | acetyl cysteine | beta-glycerophosphate |
| acetic acid | adenine | D-cysteine | creatinephosphate |
| adenosine-5-monophosphate | adenosine | D-methionine | D-glucose-6-phosphate |
| adonitol | allantoin | diethyl-dithiophosphate | diethyl-dithiophosphate |
| alpha-D-glucose | beta-phenylethylamine | DL-ethionine | DL-alpha-glycerophosphate |
| alpha-D-lactose | biuret | glutathione | potassium phosphate |
| alpha-D-melebiose | cytidine | isethionic acid | sodium pyrophosphate |
| citric acid | cytosine | L-cysteic acid | sodium thiophosphate |
| D-alanine | D-alanine | L-Cysteine | |
| D-arabinose | D-asparagine | L-djenkolic acid | |
| D-arabitol | D-aspartate | L-methionine | |
| D-asparagine | D-cysteine | magnesium sulfate | |
| D-aspartate | D-glucosamine | methane sulfonic acid | |
| D-cellubiose | D-glutamic acid | N-acetyl-DL-methionine | |
| D-cysteine | DL-alpha-amino-N-butyric acid | N-acetyl-L-cysteine | |
| D-fructose | D-methionine | potassium-tetra-thionate | |
| D-galactose | D-serine | sodium thiosulfate | |
| D-glucosamine | D-valine | sulfanic acid | |
| D-glucose | gamma-amino-N-butyric acid | taurine | |
| D-glucose-6-phosphate | glycine | taurocholic acid | |
| D-glutamate | guanidine | thiourea | |
| D-mannose | histamine | ||
| D-raffinose | inosine | ||
| D-ribose | L-alanine | ||
| D-salicin | L-arginine | ||
| D-serine | L-asparagine | ||
| D-trehalose | L-citrulline | ||
| D-xylose | L-cysteine | ||
| dulcitol | L-glutamic acid | ||
| glycerol | L-glutamine | ||
| glycine | L-glutathione | ||
| i-erythritol | L-histidine | ||
| inosine | L-isoleucine | ||
| L-alanine | L-leucine | ||
| L-arabinose | L-lysine | ||
| L-arabitol | L-methionine | ||
| L-asparagine | L-ornithine | ||
| L-aspartate | L-phenyl-alanine | ||
| L-cysteic acid | L-proline | ||
| L-cysteine | L-pyro-glutamic acid | ||
| L-fucose | L-serine; L-threonine | ||
| L-glutamic Acid | L-tryptophan | ||
| L-glutamine | L-valine | ||
| L-isoleucine | N-acetyl-D-glucosamine | ||
| L-leucine | putrescine | ||
| L-lysine | thiourea | ||
| L-methionine | thymidine | ||
| L-phenylalanine | thymine | ||
| L-pyro-glutamic acid | tyramine | ||
| L-rhamnose | tyrosine | ||
| L-serine | uridine | ||
| L-sorbose | |||
| L-threonine | |||
| L-tryptophan | |||
| L-valine | |||
| L-xylose | |||
| lactate | |||
| lactulose | |||
| malate | |||
| myo-inositol | |||
| oxalic acid | |||
| potassium sorbate | |||
| propionic acid | |||
| putrescine | |||
| quinic acid | |||
| sodium pyruvate | |||
| sodium succinate | |||
| sucrose | |||
| thymidine | |||
| xylitol |
Table 2. List of substrates used in the MAP experiments.
