In response to expression of mRNA injected into oocytes, responding animal cap tissue was assayed for expression of otx2 by in situ hybridization (Figure 2 and Table 1); otx2 is expressed in the presumptive lens ectoderm (PLE) from neural tube closure through lens placode thickening19. However, since otx2 is also expressed in the anterior neural ectoderm as well as non-neural head ectoderm outside the PLE, it is associated with both neural and placodal responses. The use of foxe3 to screen the library for a gene product capable of producing a lens-inductive response in lens-competent animal cap ectoderm allowed a more specific approach to the goal of the expression cloning, since foxe3 is expressed in the PLE from neural plate stages and throughout lens vesicle formation20, present in adjacent placodal regions but absent from neuroectoderm. Using the expression cloning and sib selection protocol above and injecting pools of library transcripts, a gene capable of producing foxe3 expression in the animal caps was isolated (Table 2). Following isolation of the clone, 179 additional animal cap assays using oocytes injected with library transcripts were screened for expression of foxe3; 50 were positive (28%). Of 140 animal cap pieces placed on uninjected oocytes, 0 were positive (Figure 3).
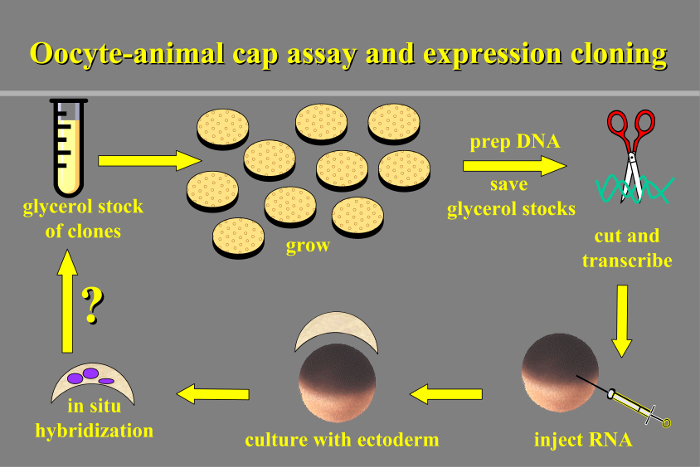
Figure 1. Oocyte-animal Cap Assay and Expression Cloning. Schematic overview of the protocol: transcripts are prepared from the clone library and injected into oocytes, animal cap ectoderm is cultured with oocytes and then assayed for induced gene expression by in situ hybridization. Please click here to view a larger version of this figure.
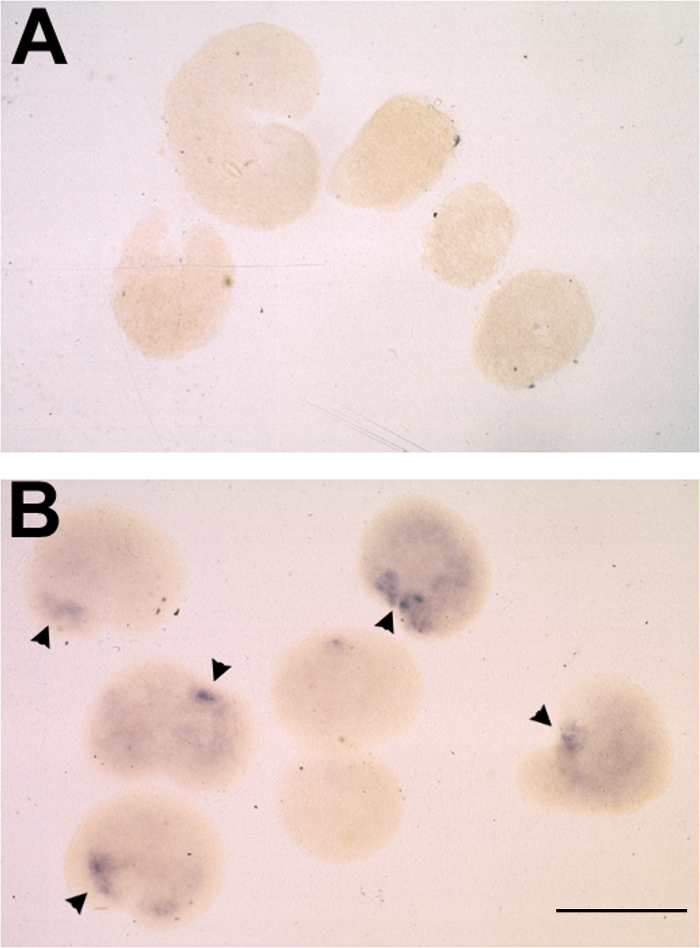
Figure 2. Typical Results of Animal Cap Assay following mRNA Injection and in situ Hybridization for Otx2. (A-B) Representative result of inductive response to dorsalized stage 14 poly(A)+ RNA in animal caps, assayed for otx2 expression by whole mount in situ hybridization. (A) Animal caps placed on uninjected oocytes at stage 10.5 and cultured to stage 25. (B) Stage 10.5 animal caps placed on oocytes injected with 10 ng RNA and cultured to stage 25. otx2 expression observed in 6/7 cases and indicated by arrowheads. Bar = 500 μm. Please click here to view a larger version of this figure.
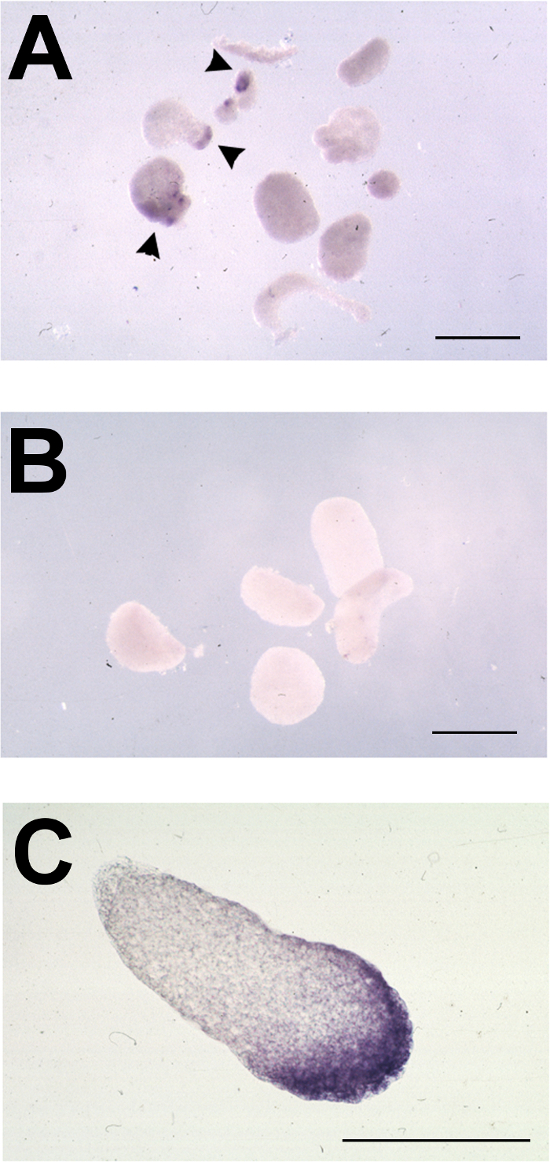
Figure 3. Typical Results of Animal Cap Assay following in situ Hybridization for Foxe3. (A-C) Representative animal caps from oocyte-animal cap assays, tested for expression of foxe3 by in situ hybridization. (A) Stage 11-11.5 animal caps placed on ldb1-injected oocytes and cultured to stage 23. foxe3 expression is indicated by arrowheads. (B) Stage 11-11.5 animal caps placed on uninjected oocytes and cultured to stage 23; no foxe3 expression detected. (C) Section through foxe3-positive induced animal cap from A showing expression in inner and outer layers of ectoderm. Bars = 500 μm. Please click here to view a larger version of this figure.
| Injected RNA | Positive cases | Gene | % |
| Stage 14 dorsalized mRNA, 10 ng | 12/24 | otx2 | 50 |
| Library pools of 105 clones, 20 ng | 3/9 | otx2 | 33 |
| Library pools of 105 to 100 clones, 20 ng | 50/179 | foxe3 | 28 |
| None | 0/140 | foxe3 | 0 |
Table 1. Oocyte-Animal Cap Assay Results. Results of animal cap assay assessed by in situ hybridization with otx2 and foxe3, using oocytes injected with mRNA or with transcripts synthesized from cDNA library pools; or uninjected oocytes.
| Injected RNA | Pool designation/selection | Positive foxe3 expression |
| Library pools of 105 clones, 20 ng | A | 2/4 |
| B* | 4/28 | |
| C | 4/44 | |
| Library pools of 104 clones, 20 ng | 1 | 0/11 |
| 2 | 0/10 | |
| 3 | 3/14 | |
| 4 | 0/12 | |
| 5 | 0/15 | |
| 6 | 1/20 | |
| 7 | 3/23 | |
| 8 | 0/16 | |
| 9 | 0/16 | |
| 10* | 5/20 | |
| Library pools of 5,000 clones, 20 ng | 1 | 0/7 |
| 2* | 5/16 | |
| 3 | 1/7 | |
| 4 | 0/7 | |
| 5 | 0/7 | |
| Library pools of 400 clones, 20 ng | 1 | 0/10 |
| 2 | 0/10 | |
| 3 | 0/10 | |
| 4 | 0/10 | |
| 5* | 8/26 | |
| 6 | 0/11 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| 9 | 0/10 | |
| 10 | 0/8 | |
| Library pools of 70-200 colonies, 20 ng | 1 | 0/8 |
| 2 | 0/10 | |
| 3* | 1/10 | |
| 4* | 1/10 | |
| 5 | 0/10 | |
| 6 | 0/9 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| Library pools of 20 colonies, 20 ng | 1 | 0/10 |
| 2 | 0/10 | |
| 3 | 0/10 | |
| 4* | 7/21 | |
| 5 | 0/10 | |
| 6 | 0/10 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| 9 | 0/10 | |
| 10 | 0/10 | |
| Library pools of 6 – 7 colonies, 20 ng | K2-6, L1 | 0/10 |
| L2-6, M7 | 0/10 | |
| M8-12, N7-8 | 0/10 | |
| K5-6, L1-4 | 1/10 | |
| L5-6, M7-10 | 0/10 | |
| M11-12, N7-8, K2-4 | 0/10 | |
| K2, K5, L2, L5, M8, M11 | 0/10 | |
| K3, K6, L3, L6, M9, M12 | 0/9 | |
| K4, L1, L4, M7, M10, N7, N8 | 1/9 | |
| Library RNAs, 20 ng | K6 | 0/10 |
| L1* | 3/10 | |
| L3 | 0/10 | |
| L4 | 0/10 | |
| Library RNA, 20 ng | L1 (ldb1) confirmation | 50/179 |
| * indicates selected pool |
Table 2. Sib Selection and Expression Cloning Results. The pool with the highest level of response in each animal cap assay (ranging between 10% and 36% positive for foxe3 expression) was selected for use in the next experiment to narrow down activity to one clone. Asterisk indicates selected pool.
