A Direct, Early Stage Guanidinylation Protocol for the Synthesis of Complex Aminoguanidine-containing Natural Products
Summary
Here, we present a protocol for direct, early stage guanidinylation that enables rapid total synthesis of aminoguanidine-containing small organic molecules. An advanced synthetic intermediate used in the synthesis of a blood coagulation factor XIa inhibitor was prepared using this protocol.
Abstract
The guanidine functional group, displayed most prominently in the amino acid arginine, one of the fundamental building blocks of life, is an important structural element found in many complex natural products and pharmaceuticals. Owing to the continual discovery of new guanidine-containing natural products and designed small molecules, rapid and efficient guanidinylation methods are of keen interest to synthetic and medicinal organic chemists. Because the nucleophilicity and basicity of guanidines can affect subsequent chemical transformations, traditional, indirect guanidinylation is typically pursued. Indirect methods commonly employ multiple protection steps involving a latent amine precursor, such as an azide, phthalimide, or carbamate. By circumventing these circuitous methods and employing a direct guanidinylation reaction early in the synthetic sequence, it was possible to forge the linear terminal guanidine containing backbone of clavatadine A to realize a short and streamlined synthesis of this potent factor XIa inhibitor. In practice, guanidine hydrochloride is elaborated with a carefully constructed protecting array that is optimized to survive the synthetic steps to come. In the preparation of clavatadine A, direct guanidinylation of a commercially available diamine eliminated two unnecessary steps from its synthesis. Coupled with the wide variety of known guanidine protecting groups, direct guanidinylation evinces a succinct and efficient practicality inherent to methods that find a home in a synthetic chemist's toolbox.
Introduction
The objective of this video is to show how using a direct and early guanidinylation method to make a terminal guanidine structure is more practical, rapid, and efficient than traditional guanidinylation methods in organic synthesis. The guanidine functional group, found on the amino acid arginine, is a key structural element in many complex natural products and pharmaceuticals. The discovery and design of new guanidine containing natural products and small molecules establish the need for a more efficient guanidinylation method. The commonly used circuitous approach features the introduction of a latent guanidine precursor that is unmasked at a late stage in the synthesis. In contrast, a straightforward tactic installs a protected guanidine onto a primary amine early in a synthetic route.
The reactive nature of guanidines generally precludes them from routine use without an appropriate protecting group strategy. Traditionally, methods to add a guanidine functional group involved an indirect approach that involved multiple protection steps followed by adding the guanidine at the end of the synthesis. Two recent syntheses illustrate the drawbacks inherent to indirect guanidinylation1,2. The direct method reported herein involves reacting a protected guanidine reagent with a primary amine early on in the synthesis of a given molecule and then deprotecting it at the end of the synthesis. This strategy was deployed successfully in recent total synthesis of biologically active marine alkaloids clavatadine A and phidianidine A and B3,4.
While this direct guanidinylation method does have its advantages over traditional methods of guanidinylation it still has its drawbacks. The chemical conditions that the protected guanidine can survive will depend on the protecting group employed. Despite these potential drawbacks, the direct guanidinylation method is an enabling strategy to add terminal guanidines to primary amines for use in the synthesis of complex organic molecules.
Protocol
Caution: Please consult and heed Safety Data Sheets (SDS) for each chemical prior to use. A few of the chemicals used in this synthesis are corrosive, toxic, carcinogenic, or otherwise harmful. Consequently, take every precaution to avoid inhalation, ingestion, or skin contact with these chemicals. Please wear appropriate Personal Protective Equipment (PPE) correctly. Proper PPE includes wrap-around safety goggles, nitrile gloves or more chemically resistant gloves, a lab coat, long pants that cover the tops of the shoes, closed-toe shoes. Use a working fume hood with the sash at the lowest possible height, in tandem with additional relevant engineering controls, to minimize the risk of accidental exposure. Portions of the procedure involve standard air- and moisture-free technique, such as the use of a Schlenk line, amber chemical storage bottles with crown caps and elastomer discs, syringe and cannula transfer of liquids and solutions, compressed gasses, and the distillation of flammable liquids under inert atmosphere.5
1. Direct Guanidinylation
- Direct guanidinylation of butane-1,4-diamine (5) with Goodman's reagent (6) to prepare di-tert-butoxycarbonyl (Boc)-agmatine (7)3,6
- Dry a 1,000 ml, three-necked, round-bottomed reaction flask containing a magnetic stir bar, a 50 ml pressure-equalizing addition funnel, and a 14/20 to 24/40 ground-glass adapter overnight in a drying oven at or above 130 °C. Remove the flask from the oven and quickly cover the aperture of the right and left necks with rubber septa. Fold the rubber septum over the lip of the flask. To the center neck, affix the glass adapter, followed by the addition funnel.
NOTE: In lieu of overnight oven drying, the flask and stir bar may be covered with a septum, placed on a Schlenk line under vacuum or inert gas, and flame-dried using a propane torch. Please consult these resources for more detailed information on using septa, a Schlenk line, and an addition funnel.7-9- Cover the top of the addition funnel with a rubber septum, folding the rubber septum over the lip of the flask. Cool the assembled apparatus to room temperature under a positive stream of inert gas using a Schlenk line.
- Place the stock bottle of butane-1,4-diamine (5) (melting point 25-28 °C) in a hot-water bath to partially melt the solid chemical. Once a small volume of liquefied butane-1,4-diamine (5) is available, use an oven-warmed Pasteur pipet to transfer 2.32 ml (2.03 g, 0.0231 mol, 3.00 molar equivalents) of compound 5 from the stock bottle to an oven-warmed 10 ml graduated cylinder. Transfer the liquefied diamine 5 to the round-bottomed reaction flask using the Pasteur pipet.
NOTE: Please consult these resources for more detailed information on using a Pasteur pipet.7,10 - Add 320 ml of dichloromethane (CH2Cl2) to the round-bottomed reaction flask from a graduated cylinder at ambient temperature (20 °C). Stir the solution while adding 1.1 ml of triethylamine (Et3N) (0.78 g, 0.0077 mol, 1.00 molar equivalents) from a 2 ml glass plunger syringe fitted with a 12-inch, 20-gauge metal needle with a beveled tip.
NOTE: Please consult these resources for more detailed information on using a syringe.7,8,10 Also consult this resource (pp. 26-29) for detailed information on using a magnetic stir plate.8 - Using an analytical balance, weigh 3.01 g (0.00769 mol, 1.00 molar equivalents) of N,N´-di-Boc-N˝-triflylguanidine (6) (Goodman's reagent).11,12 Pour this solid into a beaker. Dissolve this compound in 25 ml of anhydrous CH2Cl2. Draw this solution into a 50 ml glass plunger syringe fitted with a 12-inch, 20-gauge metal needle with a beveled tip. Dispense this solution from the syringe into the addition funnel, ensuring that the stopcock at the bottom of the funnel is closed.
NOTE: Please consult these resources for detailed information on using an analytical balance.7,13 - While continuing the magnetic stirring on the magnetic stir plate, open the stopcock slowly to allow the solution prepared in step 1.1.4 to drip into the round-bottomed reaction flask at a rate of about one drop every four seconds so that the entire solution is added over approximately 1 hr. When the addition is complete, stir the solution at ambient temperature for 12 hr. Formation of a precipitate or residue that adheres to the flask wall is typically observed throughout the 12 hr of stirring.
- Dry a 1,000 ml, three-necked, round-bottomed reaction flask containing a magnetic stir bar, a 50 ml pressure-equalizing addition funnel, and a 14/20 to 24/40 ground-glass adapter overnight in a drying oven at or above 130 °C. Remove the flask from the oven and quickly cover the aperture of the right and left necks with rubber septa. Fold the rubber septum over the lip of the flask. To the center neck, affix the glass adapter, followed by the addition funnel.
- Aqueous workup for the isolation of di-Boc-agmatine (7)
- After 12 hr, expose the solution to air by removing the septum. Remove the magnetic stir bar using a stir-bar retriever. Pour the colorless solution from the round-bottomed reaction flask into a separatory funnel.
- Wash the organic solution with two successive 50 ml portions of saturated aqueous sodium bicarbonate (NaHCO3). After each wash, drain the lower organic layer into a clean Erlenmeyer flask. Pour the upper aqueous layer into a second Erlenmeyer flask. Add the organic layer back to the separatory funnel.
NOTE: Please consult these resources for more detailed information on extraction and using a separatory funnel.7-9,14 - Wash the organic solution with two successive 50 ml portions of water. After each wash, drain the lower organic layer into a clean Erlenmeyer flask. Pour the upper aqueous layer into a second Erlenmeyer flask. Add the organic layer back to the separatory funnel.
- Wash the organic solution with one 50 ml portion of brine. Drain the lower organic layer into a clean Erlenmeyer flask, and dry with anhydrous sodium sulfate (Na2SO4). Gravity filter the solution through a funnel fitted with fluted filter paper (coarse-porosity, fast flow, 20-25 micron particle retention) into a clean, tared round-bottomed flask.
NOTE: Please consult these resources for more detailed information on drying organic solutions using a drying agent.7-9,14 Please consult these resources for more detailed information on gravity filtration.7-9,15 - Evaporate the liquid in the round-bottomed flask using a rotary evaporator with the bath temperature at 40 °C and the rotation set to 120 rpm.
NOTE: Please consult these resources for more detailed information on using a rotary evaporator.7-9,16
- Purification of di-Boc-agmatine (7)3,6
- Prepare a column for flash chromatography using an eluent of 5:3:2 ethyl acetate (EtOAc)-methanol-Et3N through a stationary phase of silica gel. Use a glass chromatography column that is 3.8 cm wide by 45 cm tall.
NOTE: Please consult these resources for more detailed information on purifying organic compounds using column chromatography.7-9,17,18- Add diatomaceous earth to the column to form a base that is approximately 2 cm tall. This filtering agent will prevent any dissolved silica gel from contaminating the column fractions. Add eluent until the column of the eluent-diatomaceous earth suspension reaches approximately 10 cm tall, about 40-50 ml. Mix the eluent and diatomaceous earth by gentle swirling to ensure the suspension is uniform, and then allow the solid to settle.
NOTE: Please consult this resource for more detailed information on using diatomaceous earth as a filtering aid.8 - Wet pack the column by making a slurry consisting of 100 g of silica gel and a sufficient volume of eluent to enable the slurry to be easily stirred using a metal spatula. Pour the slurry into the column through a plastic or glass funnel.
- Use air pressure to force the eluent through the column such that the efflux flows as a gentle stream of liquid. The rate of eluent flow should be approximately two-linear inches per minute. Stop the flow of eluent when it reaches the level of the silica gel, ensuring that the silica gel is constantly wet with the eluent.
- Add diatomaceous earth to the column to form a base that is approximately 2 cm tall. This filtering agent will prevent any dissolved silica gel from contaminating the column fractions. Add eluent until the column of the eluent-diatomaceous earth suspension reaches approximately 10 cm tall, about 40-50 ml. Mix the eluent and diatomaceous earth by gentle swirling to ensure the suspension is uniform, and then allow the solid to settle.
- Dissolve the contents of the flask (4.21 g) in a volume of the eluent that is just sufficient to dissolve the crude product completely, about 15-20 ml. Carefully load the solution onto the silica gel without disturbing the silica gel by transferring the solution from the flask to the column using a Pasteur pipet. Tap the column to ensure the top of the silica gel is flat. Drain the eluent so that it reaches the level of the silica gel. Add a small amount of the eluent and repeat.
NOTE: Please consult this resource for detailed information on dissolving solids using solvents.9- To avoid disturbing the silica gel during the elution of the column, add sand to the top of the silica gel to form a cylinder that is approximately 0.5 to 1 cm in height. Add a few milliliters of eluent to wet the sand. Drain the eluent so that it reaches the level of the sand.
- Fill the column with eluent. Use air pressure to force the eluent through the silica gel as described in step 1.3.1.3. Collect the eluent in test tubes, each test tube constituting a fraction of the eluent mixture.
- Collect fractions until the product has completely eluted from the column. To determine when this has occurred, spot one out of every few column fractions on a thin-layer chromatography (TLC) plate. Di-Boc agmatine (7) has an Rf of 0.39 in 5:3:2 EtOAc-methanol-Et3N.
NOTE: Please consult these resources for more detailed information on TLC. 7-9,19,20 - Collect all fractions containing the desired product in a tared, round-bottomed flask and evaporate the solvent using a rotary evaporator. Dry the resulting cloudy, pale yellow liquid under high vacuum overnight to remove residual solvent. Dissolve an analytical sample (approximately 5 mg) of the purified product, compound 7 (2.49 g, 98% yield), with 0.75 ml of deuterochloroform (CDCl3) and analyze it by proton (1H) and carbon (13C NMR) spectroscopy.
NOTE: Please consult these resources for more detailed information on preparing a sample for NMR analysis and conducting an NMR experiment.7-9,21
- Prepare a column for flash chromatography using an eluent of 5:3:2 ethyl acetate (EtOAc)-methanol-Et3N through a stationary phase of silica gel. Use a glass chromatography column that is 3.8 cm wide by 45 cm tall.
- Synthesis of di-Boc-agmatine isocyanate (8)3
- Prepare a clean 100 ml round-bottomed reaction flask containing a magnetic stir bar.
- On an analytical balance, add the di-Boc agmatine (7) to the round-bottomed reaction flask using a metal spatula until the added net weight of compound 7 is 1.00 g (0.00303 mol, 1.00 molar equivalents). Add 25 ml of CH2Cl2 to the round-bottomed reaction flask from a graduated cylinder at ambient temperature (20 °C). Place the solution in a water-ice bath to cool the solution to 0 °C.
- In a fume hood, use an analytical balance to weigh 0.297 g (0.00303 mol, 1.00 molar equivalents) of triphosgene. Add this solid to the round-bottomed reaction flask by pouring it through a glass or plastic funnel.
CAUTION: Triphosgene is extremely toxic. Wear two pairs of nitrile gloves when handling this compound. Its vapors are also harmful; therefore, it should only be handled in a working fume hood. Transfer an analytical balance into a working fume hood before weighing triphosgene for this reaction. - While continuing the magnetic stirring, add 25 ml of saturated aqueous NaHCO3 by pouring it into the round-bottomed reaction flask through a glass or plastic funnel. When the addition is complete, vigorously stir the biphasic mixture at 0 °C for 30 min using a magnetic stir plate.
- Aqueous workup of di-Boc-agmatine isocyanate (8)3
- After 30 min, cease stirring and remove the magnetic stir bar using a stir-bar retriever. Pour the mixture from the round-bottomed reaction flask into a separatory funnel that contains 100 ml of CH2Cl2 and 100 ml of water.
- Shake the separatory funnel vigorously to mix the layers. Drain the lower organic portion into a clean Erlenmeyer flask. Extract the aqueous layer with three successive 15 ml portions of CH2Cl2. After each extraction, drain the lower organic layer into the Erlenmeyer flask containing the combined organic extracts. After the three extractions, pour the aqueous layer into a second clean Erlenmeyer flask.
- Dry the combined organic extracts with anhydrous Na2SO4. Gravity filter the solution through a funnel fitted with fluted filter paper (coarse-porosity, fast flow, 20-25 micron particle retention) into a clean, tared 250 ml round-bottomed flask.
- Evaporate the liquid in the round-bottomed flask using a rotary evaporator with the bath temperature at 40 °C and the rotation set to 120 rpm. Dissolve an analytical sample (approximately 5 mg) of the resulting light brown oil, compound 8 (1.11 g, 103% of theoretical), in CDCl3 and analyze it by 1H and 13C NMR spectroscopy, and infrared (IR) spectroscopy.
NOTE: The isocyanate degrades within hours at room temperature, and should be used immediately in the next step upon isolation. Please consult these resources for more detailed information on infrared (IR) spectroscopy (reference 7: pp. 269-303; reference 9: pp. 146-147; reference 10: 66-76).7-9
2. Synthesis of Carbamate 9 from di-Boc Agmatine Isocyanate (8) and 2,4-DibromoHGAL (3)3
- Set up the reaction of di-Boc agmatine isocyanate (8) with 2,4-dibromoHGAL (3)
- Dry a 250 ml round-bottomed reaction flask containing a magnetic stir bar overnight in a drying oven at or above 130 °C. Remove the flask from the oven and quickly cover the flask aperture with a rubber septum. Fold the rubber septum over the lip of the flask. Cool the flask to room temperature under a positive stream of inert gas using a Schlenk line.
NOTE: In lieu of overnight oven drying, the flask and stir bar may be placed on a Schlenk line under vacuum or inert gas and flame-dried using a propane torch. - Using an analytical balance, weigh 0.933 g (0.00303 mol, 1.00 molar equivalents) of 2,4-dibromoHGAL (3), and add this solid to the round-bottomed reaction flask under a nitrogen umbrella.
NOTE: 2,4-dibromoHGAL (3) was prepared in two steps from 2,5-(dimethoxyphenyl)acetic acid (1), as shown in Figure 1a.3,22,23 The principal by-product formed during the synthesis of compound 3 is 4-bromoHGAL (4), which is formed after the first bromination of HGAL (2).3 During the purification of compound 3 by column chromatography, continued elution with 1:1 EtOAc-hexane permits collection compound 4, which has an Rf of 0.34 when the TLC eluent is 1:1 EtOAc-hexane.3 Typical isolated yields of by-product 4 range from 11-15%.3 - Add 30 ml of anhydrous CH2Cl2 to the round-bottomed reaction flask from a glass plunger syringe fitted with a 12-inch, 20-gauge metal needle with a beveled tip, at ambient temperature (20 °C). Stir to dissolve the solid.
NOTE: Distill the CH2Cl2 over calcium hydride (CaH2) under nitrogen onto activated 3 Å molecular sieves prior to use. - Add 0.103 ml of Hünig's base (0.0078 g, 0.000606 mol, 0.2 molar equivalents) to the round-bottomed reaction flask by syringe.
NOTE: "By syringe" means that the liquid was collected and dispensed using a metal/polytetrafluoroethylene-coated plunger, gas-tight syringe (glass barrel) fitted with a 6-inch, 20-gauge metal needle with a beveled tip. Distill the Hünig's base over CaH2 under nitrogen onto activated 3 Å molecular sieves prior to use. - Cover the opening of the round-bottomed flask containing the unpurified isocyanate 8 from step 1.5.4 with a rubber septum. Fold the septum over the lip of the flask. Connect the flask to the Schlenk line and purge the flask with nitrogen gas for 10 minutes.
- To the flask containing isocyanate 8, add 30 ml of anhydrous CH2Cl2 from a glass-plunger syringe fitted with a 12-inch, 20-gauge metal needle with a beveled tip at ambient temperature (20 °C). Swirl the flask to dissolve the solid.
- Dry a 250 ml round-bottomed reaction flask containing a magnetic stir bar overnight in a drying oven at or above 130 °C. Remove the flask from the oven and quickly cover the flask aperture with a rubber septum. Fold the rubber septum over the lip of the flask. Cool the flask to room temperature under a positive stream of inert gas using a Schlenk line.
- Transfer the solution of isocyanate 8 to the 2,4-dibromoHGAL (3) and Hünig's base by cannula
- Directly connect the two flasks by puncturing each septum with either beveled metal tip of an 18-inch long, 20-gauge metal cannula.
NOTE: Please consult this resource for more detailed information on using a cannula to transfer one solution to another under inert atmosphere (reference 8: pp. 74-79).8 - Remove the nitrogen inlet from the flask containing the CH2Cl2 solution of 2,4-dibromoHGAL (3) and Hünig's base, and insert a 2.5 cm disposable 16-gauge metal needle (exit needle) through the septum. Close the bubbler that serves as the exit port of the Schlenk line.
CAUTION: Closing off the bubbler of a Schlenk line that is under positive inert gas pressure can be dangerous. Ensure that the exit needle is unobstructed before closing off the bubbler. - Lower one end of the metal cannula into the CH2Cl2 solution of isocyanate 8, and transfer the entire solution to the round-bottomed reaction flask over approximately 1 hr. Adjust the nitrogen pressure as needed for the desired flow rate of approximately 0.5 ml per minute.
- Rinse the flask that contained isocyanate 8 with two successive 5 ml portions of CH2Cl2. Transfer each CH2Cl2 rinse by cannula into the round-bottomed reaction flask.
- After transferring both rinses into the reaction flask, disassemble the cannula apparatus.
- Remove the exit needle from the reaction flask while simultaneously reopening nitrogen flow to the bubbler. Transfer the nitrogen inlet originating from the Schlenk line from the flask that contained the isocyanate to the round-bottomed reaction flask.
- Remove the cannula from the round-bottomed reaction flask, and stir the solution at ambient temperature for 3 hr.
- Directly connect the two flasks by puncturing each septum with either beveled metal tip of an 18-inch long, 20-gauge metal cannula.
- Purification of carbamate 93
- After 3 hr, expose the solution to air by removing the septum. Remove the magnetic stir bar using a stir-bar retriever.
- Evaporate the liquid in the round-bottomed reaction flask using a rotary evaporator with the bath temperature at 40 °C and the rotation set to 120 rpm.
- Purify the crude product by flash column chromatography using a gradient elution of 100:0 to 90:10 CH2Cl2-diethyl ether (Et2O) through a stationary phase of silica gel. Use a glass chromatography column that is 3.8 cm wide by 45 cm tall.
- Wet pack the column by making a slurry consisting of 60 g of silica gel and a sufficient volume of CH2Cl2 to enable the slurry to be poured into the column.
- Use air pressure to force the eluent through the column such that the efflux flows as a gentle stream of liquid. The rate of eluent flow should be approximately two-linear inches per minute. Stop the flow of eluent when it reaches the level of the silica gel, ensuring that the silica gel is constantly wet with the eluent.
- Dissolve the crude product (2.202 g, 110% of theoretical) in a minimum volume of CH2Cl2. Carefully load the solution onto the silica gel without disturbing the silica gel. Tap the column to ensure the top of the silica gel is flat. Drain the eluent so that it reaches the level of the silica gel. Add a small amount of CH2Cl2 and repeat.
- To avoid disturbing the silica gel during the elution of the column, add sand to the top of the silica gel to form a cylinder that is approximately 0.5 to 1 cm in height. Add a few milliliters of CH2Cl2 to wet the sand. Drain the eluent so that it reaches the level of the sand.
- Fill the column with CH2Cl2. Use air pressure to force the eluent through the silica gel as described in step 2.3.5. Collect the eluent in test tubes, each test tube constituting a fraction of the eluent mixture.
- Collect fractions until the first compound has completely eluted from the column. To determine when this has occurred, spot one out of every few column fractions on a TLC plate.
NOTE: The first compound to elute is unreacted 2,4-dibromoHGAL (3), which has an Rf of 0.74 in 90:10 CH2Cl2-Et2O. - When the unreacted 2,4-dibromoHGAL (3) has eluted from the column, replace the eluent with 90:10 CH2Cl2-Et2O. Continue to collect fractions until the desired product has completely eluted from the column. To determine when this has occurred, spot one out of every few column fractions on a TLC plate.
NOTE: The desired product, carbamate 9, has an Rf of 0.31 in 90:10 CH2Cl2-Et2O. - Collect all fractions containing the carbamate 9 in a tared, round-bottomed flask and evaporate the solvent using a rotary evaporator. Dry the resulting foamy solid to dryness under high vacuum. Analyze an analytical sample (approximately 5 mg) of the product, carbamate 9 (1.36 g, 68% yield), by 1H and 13C NMR in CDCl3. Also collect 1H and 13C NMR spectra in deuterated dimethylsulfoxide (DMSO-d6), which permits direct solvent comparison with the product of step 3.
3. Synthesis and Isolation of Clavatadine A (10)3
- Prepare clavatadine A (10) by concomitant acid-catalyzed lactone hydrolysis and guanidine deprotection of carbamate 9
- Prepare a clean 250 ml round-bottomed reaction flask containing a magnetic stir bar.
- On an analytical balance, weigh 1.205 g (0.00181 mol, 1.00 molar equivalents) of carbamate 9, prepared in step 2, and add this solid to the round-bottomed reaction flask.
- Add 12 ml of tetrahydrofuran (THF) to the round-bottomed reaction flask from a graduated cylinder at ambient temperature (20 °C). The solid should dissolve as the solution is stirred.
- Prepare 48 ml of 1.0 M aqueous hydrochloric acid (HCl).
- Add 4 ml of concentrated (12.0 M) HCl solution to a 10 ml graduated cylinder.
- Transfer the concentrated HCl solution by Pasteur pipet to a 50 ml graduated cylinder that contains 30-40 ml of distilled water. Dilute this solution with distilled water to a final volume of 48 ml.
- Add the 48 ml of aqueous 1.0 M HCl solution prepared in step 3.1.4.3 to the round-bottomed reaction flask at ambient temperature with stirring.
- Gently place a 24/40 ground-glass stopper to cover the aperture of the reaction flask.
- Submerse the reaction flask in a water bath that has been preheated to 30 °C on a temperature-controlled hot plate. Ensure that the level of the solution in the reaction flask is below the water level in the bath.
NOTE: At this scale, the reaction will produce 2 molar equivalents of carbon dioxide and 2 molar equivalents of isobutene gas, which should occupy a space of approximately 0.174 L. Ensure the stopper is placed lightly on the flask to ensure that any excess pressure that develops can be released through the ground-glass joint. - While continuing the magnetic stirring, heat the solution at 30 °C for 20 hr.
- After 20 hr, vacuum filter the resulting suspension through a medium porosity sintered-glass funnel into a clean, tared round-bottomed flask to remove any insoluble material.
- Evaporate the yellow-colored solution in the reaction flask using a rotary evaporator with the bath temperature at 50 °C and the rotation set to 120 rpm.
- Dry the resulting yellow- to peach-colored amorphous solid to constant weight under high vacuum. Facilitate drying by heating the evacuated flask in a warm (40 °C) water bath.
- Dissolve an analytical sample (approximately 5 mg) of the product, which is the hydrochloride salt of clavatadine A (10) (0.866 g, 93% yield), in anhydrous DMSO-d6, and analyze it by one-dimensional 1H and 13C NMR spectroscopy.
Representative Results
Direct guanidinylation of a commercially available α,ω-diamine, followed by reaction with triphosgene, afforded the reactive isocyanate 8 as the linear portion of clavatadine A (Figure 1b). Yields of this two-step reaction sequence are invariably high, 95% or greater. Guanidinylation reagent 6 was prepared exactly as described by Goodman.11,24
When isocyanate 8 was combined with dibrominated phenol 3 (whose synthesis is shown in Figure 1a) in the presence of a catalytic amount of the organic base N,N-diisopropylethylamine, carbamate formation provided compound 9 (Figure 1c) in moderate yield. An aliquot of the reaction mixture was taken after 15 minutes and worked up. IR analysis of this mixture showed that the isocyanate had been fully consumed. With this data, it is unclear why the reaction yield is not higher. Reisolation of dibromophenol 3 after chromatography suggests that perhaps some of the isocyanate decomposed under the reaction conditions, or the product may have partially hydrolyzed during workup or chromatography. Finally, hydrolysis of the lactone under acidic conditions was accompanied by deprotection of the guanidine protecting groups leading to the final molecule, clavatadine A (10) (Figure 1d). Exposure of any benzofuranone-containing molecules to methanol at any stage of the synthesis invariably led to irreversible methanolysis of the lactone; therefore, contact with small alcohols is to be avoided.
Figures 2-8 include NMR or IR spectra that confirm the structure of each compound whose preparation is described herein. Comparison of the NMR spectrum of each synthesized compound with the NMR spectrum of its synthetic precursor reveals structural changes that confirm the identity of the prepared molecule. Each NMR spectrum is festooned with arrows that show likely or confirmed assignments for each spectral resonance with each group of unique hydrogen atoms in a prepared molecule. Additional supporting data that further confirms the structural assignments within synthesized molecules has been published elsewhere.3
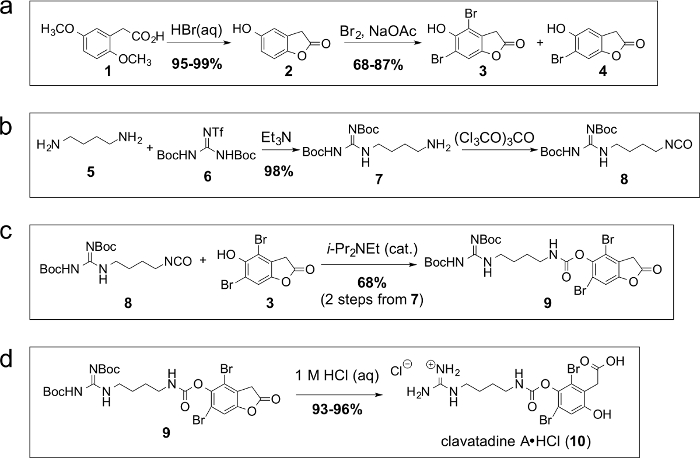
Figure 1. Step-wise synthesis of clavatadine A (10) by a direct, early stage guanidinylation approach. Schemes a-d illustrate the sequence of chemical reactants and reaction conditions for the preparation of clavatadine A (10) by a direct, early stage guanidinylation approach. (a) Synthesis of the aromatic portion, 2,4-dibromohomogentisic acid lactone (3). (b) Synthesis of the linear portion, isocyanate 8, by direct guanidinylation. (c) Carbamate-forming reaction uniting the aromatic and linear subunits. (d) Acidic hydrolysis and Boc-deprotection of carbamate 9 leading to the hydrochloride salt of clavatadine A (10). Please click here to view a larger version of this figure.
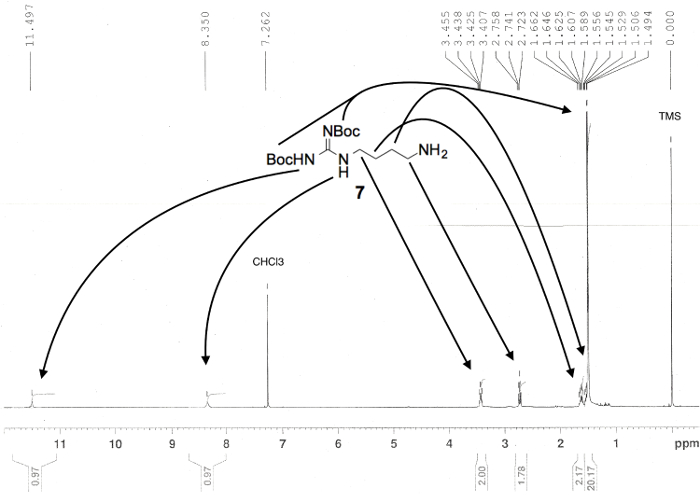
Figure 2. Proton NMR spectrum confirming the preparation of linear compound di-Boc-agmatine (7). Numerical values pertaining to chemical shifts and relative integration of visible proton signals are labeled above and below the spectrum, respectively; peak assignments originate from the structure shown; and known impurities are listed above each relevant peak3. Please click here to view a larger version of this figure.
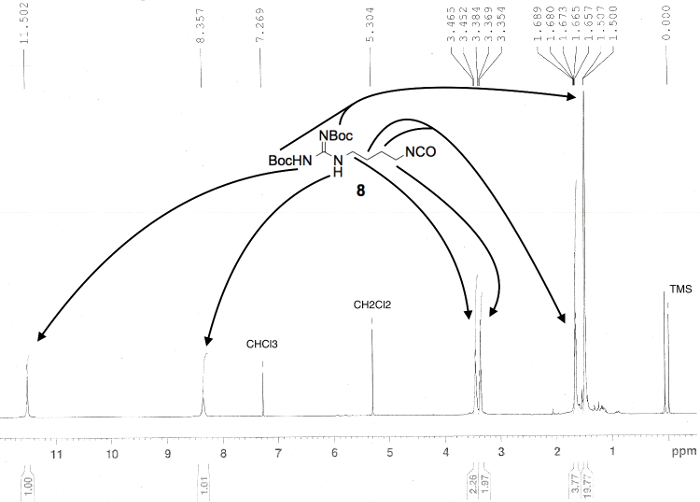
Figure 3. Proton NMR spectrum confirming the preparation of linear compound di-Boc-agmatine isocyanate (8). Numerical values pertaining to chemical shifts and relative integration of visible proton signals are labeled above and below the spectrum, respectively; peak assignments originate from the structure shown; and known impurities are listed above each relevant peak3. Please click here to view a larger version of this figure.
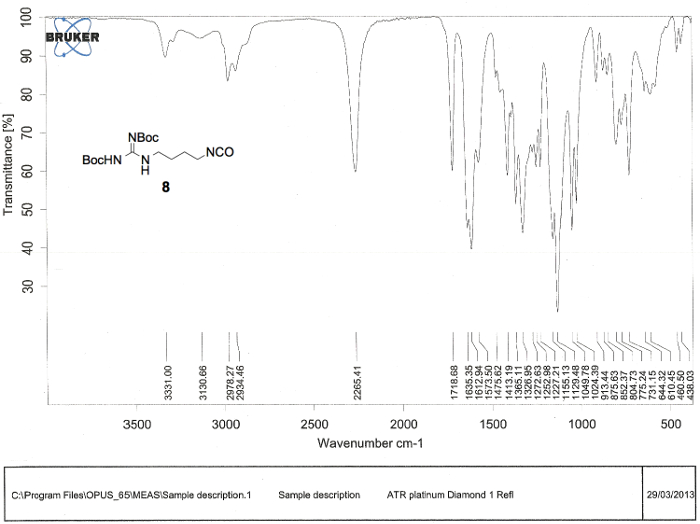
Figure 4. Infrared (IR) spectrum confirming the preparation of linear compound di-Boc-agmatine isocyanate (8). Numerical values pertaining to wave numbers of bond absorptions are labeled below the spectrum, but above the abscissa. The characteristic isocyanate stretch of compound 8 can be found at 2,265 cm-1. Please click here to view a larger version of this figure.
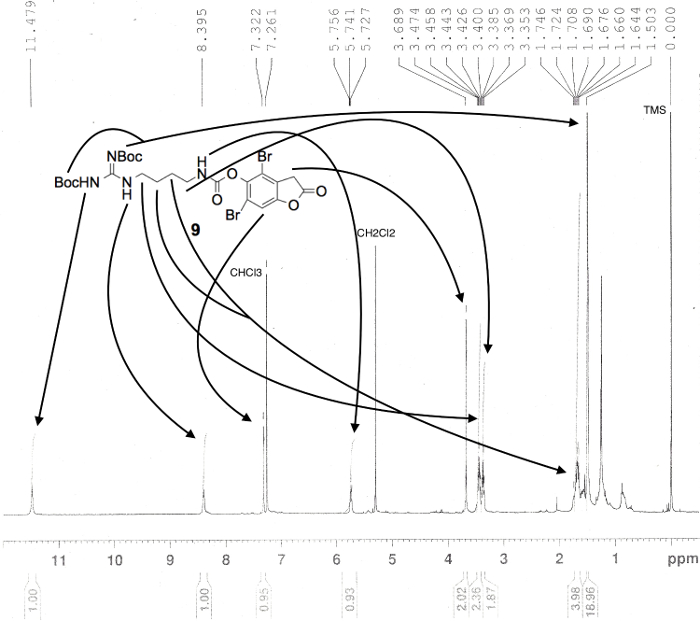
Figure 5. Proton NMR spectrum confirming the preparation of carbamate 9, in CDCl3. Numerical values pertaining to chemical shifts and relative integration of visible proton signals are labeled above and below the spectrum, respectively; peak assignments originate from the structure shown; and known impurities are listed above each relevant peak3. Please click here to view a larger version of this figure.
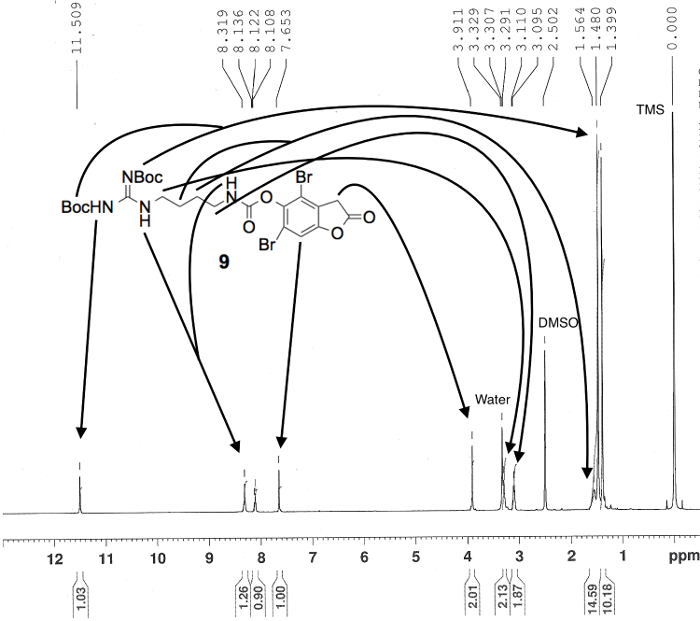
Figure 6. Proton NMR spectrum confirming the preparation of carbamate 9, in DMSO-d6. Numerical values pertaining to chemical shifts and relative integration of visible proton signals are labeled above and below the spectrum, respectively; peak assignments originate from the structure shown; and known impurities are listed above each relevant peak. Please click here to view a larger version of this figure.
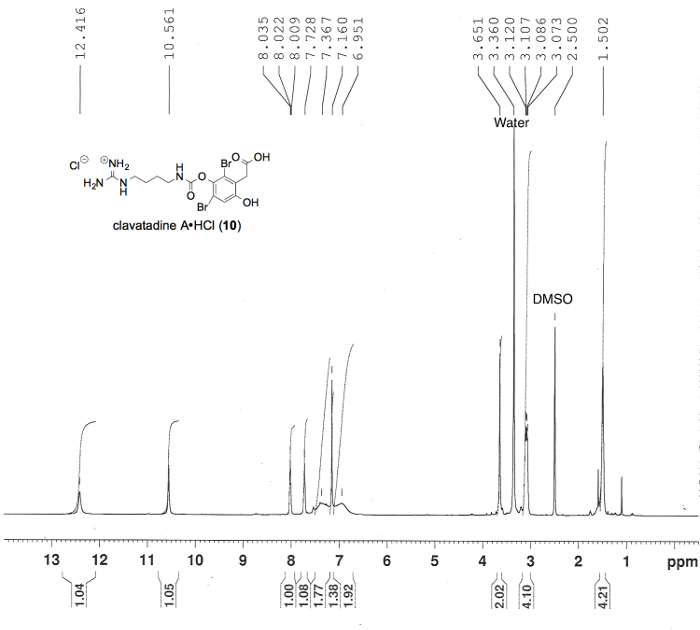
Figure 7. Proton NMR spectrum confirming the preparation of clavatadine A (10), in DMSO-d6. Numerical values pertaining to chemical shifts and relative integration of visible proton signals are labeled above and below the spectrum, respectively; peak assignments originate from the structure shown; and known impurities are listed above each relevant peak3. Please click here to view a larger version of this figure.
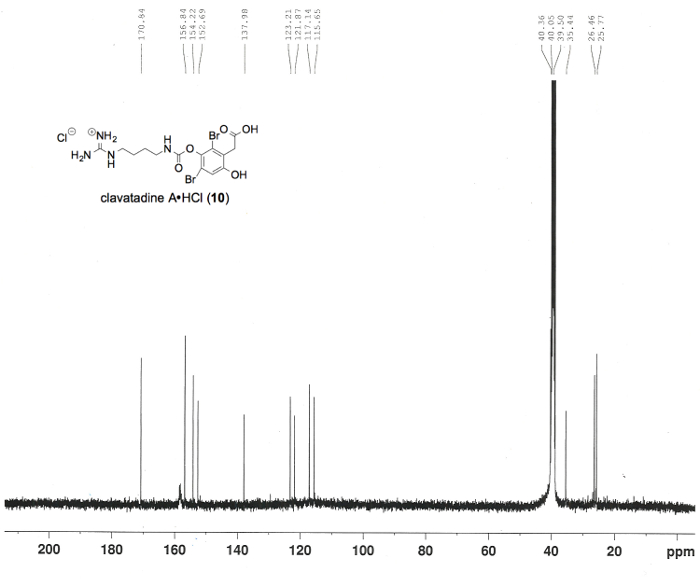
Figure 8. Carbon (13C) NMR spectrum confirming the preparation of clavatadine A (10), in DMSO-d6. Numerical values pertaining to chemical shifts are labeled above the spectrum3. Please click here to view a larger version of this figure.
Discussion
Initial efforts to prepare clavatadine A enlisted a traditional, indirect approach to guanidinylation from a suitable amine precursor, which in this case was a terminal azide. Central to this effort was the union of the two halves of the molecule to construct the carbamate moiety. Unfortunately, all attempts to realize an azide reduction in anticipation of a planned late-stage guanidinylation were unsuccessful.25,26 These setbacks inspired the pursuit of compound 7, which could be prepared in a single step by direct guanidinylation from commercially available materials. Though this method had been used in prior total syntheses, in this instance, a direct approach circumvented a critical impasse encountered amid myriad attempts to install the protected guanidine functionality into an advanced synthetic intermediate.27-29
The application of this direct aminoguanidinylation approach began with the preparation of the known N,N´-di-Boc-protected guanidine 7 from Goodman's reagent (6) and 1,4-butanediamine (5) in high yield.3,6,30 The terminal amine of protected guanidine 7 was converted into the reactive isocyanate 8 by exposure to triphosgene in a bi-phasic solvent mixture.3 In the penultimate synthetic transformation, the electrophilic isocyanate 8 was treated with 2,4-dibromohomogentisic acid lactone (3) to form the central carbamate linkage in compound 9.3 Finally, tandem lactone hydrolysis and guanidine deprotection occurred under dilute acidic conditions to provide clavatadine A (10) in 93% yield.3 The overall yield for the entire four-step synthesis (longest linear sequence) is 41-43%.3
For each chemical reaction described in the protocol that was not conducted in aqueous media, the use of high-purity, moisture-free solvents was critical. Some of the reactive intermediates formed during these transformations likely react with adventitious water, leading to decomposition. Although Goodman's reagent (6) is commercially available, its substantial cost and relative ease of synthesis made its preparation a reasonable choice. Again, minimizing moisture by distilling each reagent and painstaking temperature control were critical to its successful synthesis, as stated in the published procedure.11
Despite the expediency inherent to this direct approach for the synthesis of biologically relevant aminoguanidine-containing compounds, there are limitations to this method. Using different amine protection groups is possible in this direct guanidinylation method, but the overall success will always depend on the chosen protecting group strategy. Principally, the selection of aminoguanidine protecting groups requires substantial forethought, because the masked guanidine must remain intact throughout every subsequent synthetic step. In addition, the advanced target molecule must be able to endure the conditions and reagents required for guanidine deprotection at the appropriate time. In the synthesis of clavatadine A (10), acid-sensitive Boc groups were used to protect the guanidine, which necessitated the avoidance of reactions that required or created an acidic environment. In this case, the need to employ acidic conditions to hydrolyze the lactone was optimal due to the fact that acid is a convenient means to cleave Boc carbamates.31 Although clavatadine A represented an ideal template to showcase this approach, direct guanidinylation should be amenable to the preparation of many other natural and non-natural organic molecules. To this end, efforts are underway in our laboratory to prepare several non-natural analogues of clavatadine A as part of a drug-discovery program to develop a reversible and selective, natural-product based inhibitor of human blood coagulation factor XIa.32
What makes this direct method potentially better than the traditional, indirect approach is that it can shorten an organic synthesis route by multiple steps, removing the need to protect and deprotect a terminal amine multiple times before installing the desired guanidine functionality. Though traditional, indirect guanidinylation methods are effective, such as that illustrated in Looper's recent total synthesis of saxitoxin, the inclusion of extraneous steps in a synthesis is time-intensive and can potentially lower the overall yield.33 Moreover, the value of direct guanidinylation was highlighted in a recent total synthesis of 1,2,4-oxadiazole-containing natural products phidianidine A and B. This total synthesis was two steps shorter than the synthesis reported one year earlier by Snider and co-workers.4,34
In the future, the direct guanidinylation method needs to be expanded and tested on different aminoguanidine-containing scaffolds, inescapably, toward the exploration of varied guanidine protecting groups. Clavatadine A and phidianidine A and B both used Boc protecting groups to mask the guanidine functionality. The next stage in the refinement of this method would be to try the same reactions with different protecting groups to see if higher yields can be obtained.4 Recent work by Pfeffer,35 Looper,36 and Nagasawa37 suggests that a variety of aminoguanidine protecting groups in addition to Boc, such as Cbz, as well as derivatives of Cbz may be enlisted. Another approach would involve the use of two different protecting groups on the aminoguanidine scaffold. Judiciously chosen masking groups with orthogonal reactivity may enable the aminoguanidine to survive reaction conditions that cleave one protecting group while leaving the other intact.38 In conclusion, the direct guanidinylation method used for the total synthesis of clavatadine A and variations thereof may be used to synthesize newly discovered guanidine-containing natural products and designed pharmaceuticals.39,40
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Dr. John Greaves and Ms. Soroosh Sorooshian, Department of Chemistry, University of California, Irvine Mass Spectrometry Facility, for mass spectrometric analyses. We also thank Mr. Jacob Buchanan for helpful discussions, as well as Miss Stephanie J. Conn, Mrs. Shannon M. Huffman (Vreeland), and Miss Alexandra N. Wexler for early stage work on this project. Partial funding was provided by the Central Washington University (CWU) School of Graduate Studies (C.E.M), the CWU Seed Grant Program, and the CWU Faculty Research Program.
Materials
| Chloroform-d | Sigma-Aldrich | 612200-100G | 99.8% D, 0.05% v/v tetramethylsilane, Caution: toxic |
| Dimethylsulfoxide-d6 | 185965-50G | 99.9% D, 1% v/v tetramethylsilane | |
| sodium thiosulfate pentahydrate | Sigma-Aldrich | S8503-2.5KG | |
| sodium sulfate, anhydrous | Sigma-Aldrich | 238597-2.5KG | |
| silica gel | Fisher Scientific | S825-25 | Merck, Grade 60, 230-400 mesh |
| washed sea sand | Sigma-Aldrich | 274739-5KG | |
| hexane | Sigma-Aldrich | 178918-20L | Caution: flammable |
| ethyl acetate | Sigma-Aldrich | 319902-4L | |
| methylene chloride | Sigma-Aldrich | D65100-4L | |
| sodium chloride | Sigma-Aldrich | S9888-10KG | |
| sodium bicarbonate | Sigma-Aldrich | S6014-2.5KG | |
| acetic acid | Sigma-Aldrich | 695092-2.5L | |
| hydrochloric acid | Sigma-Aldrich | 258248-2.5L | Caution: Corrosive |
| bromine | Sigma-Aldrich | 470864-50G | >99.99% trace metals basis Caution: Corrosive, causes severe burns |
| hydrobromic acid | Sigma-Aldrich | 244260-500ML | 48% aqueous, Caution: Corrosive |
| 2,5-dimethoxyphenylacetic acid | ChemImpex | 26909 | |
| chloroform | Sigma-Aldrich | 132950-4L | Caution: Toxic |
| tetrahydrofuran | Sigma-Aldrich | 360589-4x4L | Caution: highly flammable |
| N,N-diisopropylethylamine | Sigma-Aldrich | D125806-500ML | Caution: Corrosive |
| triethylamine | Sigma-Aldrich | T0886-1L | Caution: Corrosive |
| 3 Angstrom molecular sieves | Sigma-Aldrich | 208574-1KG | |
| calcium hydride | Sigma-Aldrich | 213268-100G | Caution: Corrosive, reacts violently with water |
| ammonium molybdate | Sigma-Aldrich | 431346-50G | |
| phosphomolybdic acid | Sigma-Aldrich | 221856-100G | |
| cerium (IV) sulfate | Sigma-Aldrich | 359009-25G | |
| 1-butanol | Sigma-Aldrich | 537993-1L | |
| 1,4-butanediamine | Sigma-Aldrich | D13208-100G | Caution: Corrosive / warm in hot water bath to melt prior to use |
| triphosgene | VWR | 200015-064 | Caution: Highly Toxic |
| methanol | Sigma-Aldrich | 646377-4X4L | |
| sodium acetate | Sigma-Aldrich | 241245-100G | |
| Dimethylsulfoxide-d6 | Sigma-Aldrich | 570672-50G | Anhydrous, 99.9% D |
| sodium hydroxide | Sigma-Aldrich | 221465-500G | Caution: Corrosive |
| guanidine hydrochloride | Sigma-Aldrich | G4505-25G | Caution: Toxic, Corrosive |
| di-tert-butyl dicarbonate | VWR | 200002-018% | Caution: Toxic / may warm in hot water bath to melt prior to use |
| trifluoromethanesulfonic anhydride | Fisher Scientific | 50-206-771 | 98%, anhydrous, Caution: toxic, corrosive, extremely moisture sensitive |
References
- Adabala, P. J. P., Legresley, E. B., Bance, N., Niikura, M., Pinto, B. M. Exploitation of the Catalytic Site and 150 Cavity for Design of Influenza A Neuraminidase Inhibitors. J. Org. Chem. 78 (21), 10867-10877 (2013).
- Trost, B. M., Kaneko, T., Andersen, N. G., Tappertzhofen, C., Fahr, B. Total Synthesis of Aeruginosin 98B. J. Am. Chem. Soc. 134 (46), 18944-18947 (2012).
- Conn, S. J., Vreeland, S. M., Wexler, A. N., Pouwer, R. H., Quinn, R. J., Chamberland, S. Total Synthesis of Clavatadine. A. J. Nat. Prod. 78, 120-124 (2014).
- Buchanan, J. C., Petersen, B. P., Chamberland, S. Concise Total Synthesis of Phidianidine A and B. Tetrahedron Lett. 54, 6002-6004 (2013).
- . . Technical Bulletin AL-134: Handling Air-Sensitive Reagents. , (2012).
- Castagnolo, D., Raffi, F., Giorgi, G., Botta, M. Macrocyclization of Di-Boc-guanidino-alkylamines Related to Guazatine Components: Discovery and Synthesis of Innovative Macrocyclic Amidinoureas. Eur. J. Org. Chem. 2009 (3), 334-337 (2009).
- Zubrick, J. W. . The Organic Chem Lab Survival Manual: A Student’s Guide to Techniques. , (2012).
- Pirrung, M. C. . The Synthetic Organic Chemist’s Companion. , (2007).
- Padias, A. B. . Making the Connections 2: A How-To Guide for Organic Chemistry Lab Techniques, Second Edition. , (2011).
- . . Digital Lab Techniques Manual: Volumetric Techniques. , (2007).
- Baker, T. J., Tomioka, M., Goodman, M. Preparation and Use of N,N’-Di-Boc-N"-Triflylguanidine. Org. Synth. 78, 91-98 (2002).
- Baker, T. J. Synthesis of Biologically Important Guanidine-Containing Molecules Using Triflyl-Diurethane Protected Guanidines. Synthesis. S1, 1423-1426 (1999).
- . . Digital Lab Techniques: Using a Balance. , (2007).
- . . Digital Lab Techniques: Reaction Work-Up I: Extraction, Washing, and Drying. , (2007).
- . . Digital Lab Techniques: Filtration. , (2007).
- . . Digital Lab Techniques: Reaction Work-Up II: Using the Rotovap. , (2007).
- . . Digital Lab Techniques: Column Chromatography. , (2007).
- . . Flash Chromatography 101. , (2015).
- . . Digital Lab Techniques Manual: TLC – The Basics. , (2007).
- . . Digital Lab Techniques: TLC – Advanced. , (2007).
- Massachusetts Institute of Technology Department of Chemistry Instrumentation Facility. . NMR Tips and Tricks. , (2015).
- Krohn, K. Synthese des Bactereostatischen 2.4-Dibrom-homogentisinsäure – Amids und Verwandter Verbindungen. Tetrahedron Lett. 16 (52), 4667-4668 (1975).
- Wolkowitz, H., Dunn, M. S. Homogentisic Acid. Biochem. Prep. 4, 6-11 (1955).
- Feichtinger, K., Zapf, C., Sings, H. L., Goodman, M. Diprotected Triflylguanidines: A New Class of Guanidinylation Reagents. J. Org. Chem. 63, 3804-3805 (1998).
- Ariza, X., Urpì, F., Vilarrasa, J. A practical procedure for the preparation of carbamates from azides. Tetrahedron Lett. 40, 7515-7517 (1999).
- Ariza, X., Urpì, F., Viladomat, C., Vilarrasa, J. One-Pot Conversion of Azides to Boc-Protected Amines with Trimethylphosphine and Boc-ON. Tetrahedron Lett. 39, 9101-9102 (1998).
- Snider, B. B., Song, F., Foxman, B. M. Total Syntheses of (±)-Anchinopeptolide D and (±)-Cycloanchinopeptolide. D. J. Org. Chem. 65 (3), 793-800 (2000).
- Barykina, O., Snider, B. B. Synthesis of (+-)-Eusynstyelamide A. Org. Lett. 12 (11), 2664-2667 (2010).
- Yu, M., Pochapsky, S. S., Snider, B. B. Synthesis of (±)-Bistellettadine A. Org. Lett. 12 (4), 828-831 (2010).
- Expòsito, A., Fernández-Suárez, M., Iglesias, T., Muñoz, L., Riguera, R. Total Synthesis and Absolute Configuration of Minalemine A, a Guanidine Peptide from the Marine Tunicate Didemnum rodriguesi. J. Org. Chem. 66, 4206-4213 (2001).
- Wuts, P. G. M., Greene, T. W. . Greene’s Protective Groups in Organic Synthesis. , (2007).
- Malmberg, C. E., Chamberland, S. Total synthesis of clavatadine A analogues to produce a viable reversible inhibitor for factor XIa. , (2015).
- Bhonde, V. R., Looper, R. E. A Stereocontrolled Synthesis of (+)-Saxitoxin. J. Am. Chem. Soc. 133, 20172-20174 (2011).
- Lin, H. Y., Snider, B. B. Synthesis of Phidianidines A and B. J. Org. Chem. 77, 4832-4836 (2012).
- Hickey, S. M., Ashton, T. D., Pfeffer, F. M. Facile Synthesis of Guanidine Functionalised Building Blocks. Asian J. Org. Chem. 4 (4), 320-326 (2015).
- Looper, R. E., Haussener, T. J., Mack, J. B. C. Chlorotrimethylsilane Activation of Acylcyanamides for the Synthesis of Mono-N-acylguanidines. J. Org. Chem. 76, 6967-6971 (2011).
- Shimokawa, J., Ishiwata, T., et al. Total Synthesis of (+)-Batzelladine A and (+)-Batzelladine D, and Identification of Their Target Protein. Chem. Eur. J. 11, 6878-6888 (2005).
- Katritzky, A. R., Rogovoy, B. V. Recent Developments in Guanylating Agents. ARKIVOC. iv, 49-87 (2005).
- Berlinck, R. G. S., Trindade-Silva, A. E., Santos, M. F. C. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 29, 1382-1406 (2012).
- Ebada, S., Proksch, P. Chemical and Pharmacological Significance of Natural Guanidines from Marine Invertebrates. Mini-Rev. Med. Chem. 11 (3), 225-246 (2011).
