For construction of a new VV vector, the key starting point is to design and construct a donor shuttle vector that can target a particular region of the genome. In this study, the VV N1L gene was used as an example target. The cassette of the donor shuttle vector for the recombination and the targeted region in the VV are shown in Figure 1. In order to enhance the efficiency of the homologous recombination, plasmids expressing Cas9 and a N1L-specific gRNA were co-transfected into the CV-1 cells 48 h prior to performing the homologous recombination33. One day (24 h) post-transfection, RFP was expressed in the CV-1 cells (Figure 2). After infection of CV-1 cells with the whole lysate from the homologous recombination (step 5), RFP-positive and negative plaques were both observed in the CV-1 cells (Figure 3). Following the purification protocol described, a pure plaque could be obtained after 3-5 rounds of purification. As shown in Figure 4, all plaques after infection with cell lysate derived from a pure plaque with the mutant vaccinia virus presented an RFP signal. To verify whether the pure mutant VV had the correct gene modification, PCR was used to confirm the absence of the targeted N1L gene, as shown in Figure 5. PCR of the modified virus showed a positive signal for RFP and an absent signal for N1L (Figure 5 lane A-G), while the PCR results of N1L for the control virus (Ctr) with an intact N1L region is positive. The control DNA fragments of A52R were positive for all recombinant viruses and the Ctr virus. The HR efficiency in RFP-positive plaques is 100% (6/6) in this experiment (it was up to 94% in the previous work34). The method described herein has improved the recombination efficiency by more than 100 fold compared to conventional protocols33, 34.
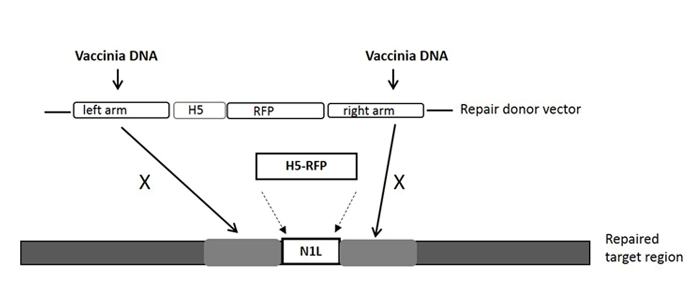
Figure 1: The cassette of the donor shuttle vector for the homologous recombination, and the targeted region in the VV genome. The purification marker gene RFP is driven by the H5 promoter. The repair donor vector targeting the N1L region results in the RFP gene being incorporated into the N1L region after homologous recombination. 'X' indicates the homologous sequence in the repair donor vector that will replace the same sequence on the vaccinia virus genome. Please click here to view a larger version of this figure.
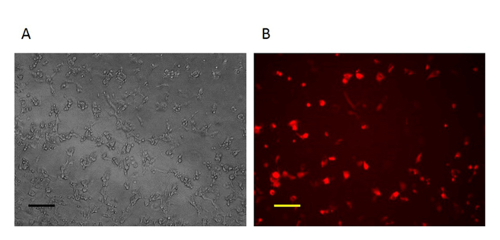
Figure 2: Imaging of the CV-1 cells one day after homologous recombination. One day after homologous recombination, the cells infected with VV and transfected by repair donor vector are RFP-positive. A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
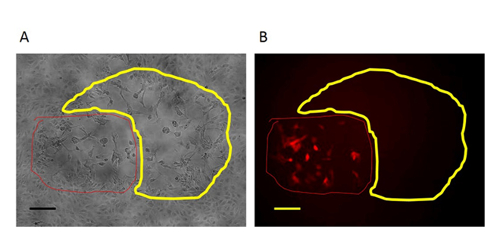
Figure 3: An RFP-positive plaque in the first round purification of mutant virus. In the first-round purification of the mutant virus, the RFP-positive plaque (circled with red line) with the target region deletion are surrounded by plaques formed by wild type virus (circled with yellow line). A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
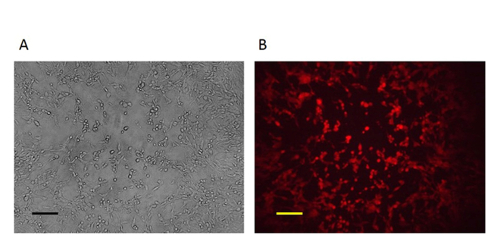
Figure 4: The pure mutant VV. After 3-5 rounds of purification, pure plaques were obtained as all the plaques were RFP-positive. A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 5: Verification of the pure mutant VV. DNA was extracted from VV infected CV-1 cells. The deletion of the target region N1L was verified by PCR using N1L primers, the insertion of RFP into N1L region was confirmed by PCR using RFP primers. Lanes A-F correspond to six purified plaques, lane G is a control plaque with N1L deletion verified in previous work33, lane Ctr (control) is the wild type vaccinia virus (with N1L region intact). A52R gene was amplified as the control gene for all the samples. Please click here to view a larger version of this figure.
