In 1998, Fire and Mello reported that double-stranded RNA (dsRNA) can induce inhibition of gene function in Caenorhabditis elegans1. This response triggered by dsRNA was named RNA interference (RNAi), and such RNAi-mediated gene silencing was reported to be conserved in animals, plants, and fungi2-7. In plants and some animals, RNAi functions systemically, meaning that the effect can spread to other cells/tissues where dsRNA is not directly introduced (reviewed in8-10). Scientists have made use of this endogenous cellular RNAi response by designing dsRNAs to target genes of interest, thereby knocking down gene function without directly manipulating the genome (reviewed in11-14).
RNAi is a powerful tool for functional studies due to the following advantages. First, even with minimal gene sequence information, a gene can be targeted using RNAi. This is especially important for studies of non-model organisms lacking genomic or transcriptomic data. Second, in organisms where the RNAi response is robustly systemic, RNAi-mediated gene knockdown can be performed at almost any developmental stage. This feature is very useful for studying the function of pleiotropic genes. Third, in some cases, RNAi effects spread to the gonads and progeny, such that phenotypes are observed in offspring15,16. This phenomenon, known as parental RNAi (pRNAi), is especially advantageous for genes impacting embryonic development, as numerous offspring produced by a single injected parent can be examined without direct manipulation of eggs. For these reasons, pRNAi is the method of choice. However, if pRNAi is ineffective, for example for genes required for oogenesis, then embryonic RNAi (eRNAi) must be used. Fourth, RNAi can be used to generate the equivalent of an allelic series in that the amount of dsRNA delivered can be varied over a range to produce weak to strong defects. Such a gradation of phenotypes can be helpful for understanding gene function when the gene is involved in a complex process and/or complete loss of function is lethal. Fifth, delivery of dsRNA is generally easy and feasible, especially in animals showing robust systemic RNAi responses. dsRNA can be introduced by microinjection1,5, feeding/ingestion17,18, soaking,19,20 and virus/bacteria-mediated delivery21,22. Sixth, unlike some gene targeting/editing methods, there is no need to screen for organisms carrying the mutation or to carry out genetic crosses to generate homozygotes when using RNAi. Therefore, compared to many other techniques for studying gene function, RNAi is fast, inexpensive, and can be applied for large-scale screens23-25.
The broad utility of RNAi provides means to carry out functional studies in a wide range of organisms, expanding the range of species available for study beyond the traditional model systems for which genetic tools have been developed. For example, studies using non-model systems are required to give insights into the evolution of genes and gene networks by comparing the functions of orthologs from species representing different development modes or exhibiting distinct morphological features26-29. These types of studies will provide a better understanding of biological diversity, with impacts for both applied and basic research.
Being the largest animal group on the planet, insects provide a great opportunity to explore the mechanisms underlying diversity. Additionally, insects are generally small, have short life cycles, high fecundity, and are easy to rear in the lab. In the past two decades, RNAi has been successfully applied in insects spanning orders, including Diptera (true flies)5, Lepidoptera (butterflies and moths)30, Coleoptera (beetles)16,31, Hymenoptera (sawflies, wasps, ants and bees)32, Hemiptera (true bugs), Isoptera (termites)34, Blattodea (cockroaches)35, Orthoptera (crickets, grasshoppers, locusts, and katydids)36and Phthiraptera (lice)37. Successful application of RNAi has provided functional data for studies of patterning in early embryogenesis (anterior-posterior axis32, dorsal-ventral axis28, segmentation26,38), sex determination39,40, chitin/cuticle biosynthesis41, ecdysone signaling42, social behavior43, and more. RNAi methods developed for different insect species may be of additional benefit in that they are likely to be useful for pest control (reviewed in44-46). RNAi effects will be gene-specific as well as species-specific, as long as non-conserved regions are chosen for targeting. For beneficial insect species like honeybees and silkworms, targeting genes vital for the survival of viruses or parasites to control infection may provide a novel strategy to protect these species47,48.
Dermestes maculatus (D. maculatus), common name hide beetle, is distributed worldwide except for Antarctica. As a holometabolous insect, the D. maculatus life cycle includes embryonic, larval, pupal, and adult stages (Figure 1). Because it feeds on flesh, D. maculatus is used in museums to skeletonize dead animals and forensic entomologists can use it to estimate time of death49,50. D. maculatus feeds on animal products including carcasses, dried meat, cheese, and the pupae/cocoons of other insects and thus causes damage to households, stored food, and the silk, cheese, and meat industries 51,52. Applying RNAi in this beetle could provide an efficient and environmentally friendly way to minimize its economic impact. Our lab has used D. maculatus as a new model insect to study segmentation53. In addition to being amenable to lab rearing, D. maculatus is of interest for basic research as it is an intermediate-germ developer, making it a useful species to study the transition between short- and long-germ development.
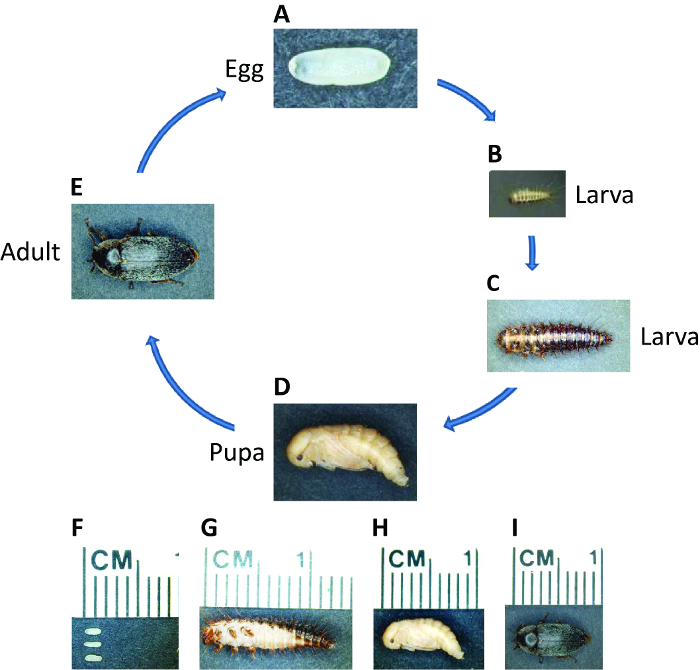
Figure 1: Life Cycle of D. maculatus. Photographs of D. maculatus at different life stages, as indicated. The life cycle from egg to adult takes three weeks at 30 °C but longer at lower temperatures. (A, F) Freshly laid embryos are white to light yellow and oval, approximately 1.5 mm in length. Embryogenesis takes ~55 hr at 30 °C. (B, C and G) Larvae have dark pigmented stripes and are covered with setae. Larvae go through several instars depending on the environment and their length can extend up to over 1 cm. (D, H) Young pupae are light yellow. Pupation takes ~ 5 – 7 days at 30 °C. (E, I) Shortly after eclosion, dark pigmentation appears over the adult beetle body. Adults can live up to several months and one female can lay hundreds of embryos over her lifetime. Please click here to view a larger version of this figure.
Previously, we showed that RNAi is effective in knocking down gene function in D. maculatus53. Here our experience rearing D. maculatus colonies in the laboratory is shared along with step-by-step protocols for both embryonic and parental RNAi set-up, injection, post-injection care, and phenotypic analysis. The dsRNA-mediated gene knockdown and analysis methods introduced here not only provide detailed information for addressing questions in D. maculatus, but also have potential significance for applying RNAi in other non-model beetle/insect species.
1. Rearing of D. maculatus
NOTE: A breeding colony of D. maculatus was set up in the authors' lab using adults and larvae purchased commercially. The species identity was verified using DNA barcoding53.
- To set up a new cage in the lab, spread a thin layer of wood shavings into a medium-size insect cage (30.5 x 19 x 20.3 cm3). Place a ~ 10 x 6 x 3 cm3 chunk of Styrofoam in the cage to let larvae hide for pupation. Add 20 – 50 beetles (either adults or late instar larvae). Beetles will hide in the wood shavings.
- Add wet cat food in a Petri dish or a weighing boat. Cover the cage with mesh cloth and place the cage in an incubator.
- For maintaining a breeding colony in the lab, set the temperature between 25 and 30 °C. D. maculatus grows faster at higher temperatures, but this also promotes the growth of fungus and mites. To maintain a healthy colony, use 25 – 28 °C for regular stock maintenance and 30 °C for rapidly expanding the colony. The life cycle takes approximately three weeks to four months depending on environmental factors50 (Figure 2).

Figure 2: D. maculatus Lab Colony. Photograph of a typical D. maculatus insect cage is shown. Wood shavings are spread to let the beetles hide. Cat food is added in a small Petri dish or weighing boat. Styrofoam is placed into the cage as a refuge for final instar larvae to pupate. The cage shown here is 30.5 x 19 x 20.3 cm3 and houses a few hundred larvae, pupae, and adults. Please click here to view a larger version of this figure.
- Leave eggs in the cage to mature. To expand the colony, establish new cages with eggs (collected as described in Section 2), larvae, or adults, as above. D. maculatus lay eggs throughout the cage, especially near the food source.
- Replace wet cat food about twice a week.
NOTE: Due to the unpleasant odor of wet cat food, alternative food sources were also tested (see Discussion).
2. Embryo Collection
- To set up a collection, separate at least 50 males and 50 females from the colony. Young adults are best. Place adults in a mini-sized cage (17.8 x 10.2 x 12.7 cm3) without wood shavings. Add cat food in a weighing boat.
- Before starting collection, check the cage carefully for any embryos present and remove them. Put a stretched cotton ball into the cage, and leave the cage in a 30 °C incubator. After the appropriate time window, the cotton ball will be ready for embryo collection.
- Fold a piece of black construction paper (A4 size) in half to create a crease, and then unfold.
- Remove the stretched cotton ball from the cage. Remove adults from the cotton ball and put them back into the cage.
- While pinching the stretched cotton ball very gently, tear it apart slowly into thin cotton filaments to let the eggs fall onto the black paper.
NOTE: D. maculatus embryos are fragile and hard to see in cotton. They can be easily crushed if holding the cotton ball too firmly.
3. dsRNA Preparation
- DNA Template Preparation for dsRNA Synthesis
- Run 4 – 6 50 µl PCR reactions using primers containing T7 promoter sequences at both 5' ends to amplify DNA template according to manufacturer's protocol.
- Purify PCR product using a commercial PCR purification kit, following manufacturer's instructions. The ideal final concentration of DNA template is ≥ 100 ng/µl.
- Injection Buffer Preparation
- Prepare 100 ml of injection buffer (0.1 mM NaH2PO4, 5 mM KCl). Adjust pH to 6.8 with 5 M NaOH.
- Store aliquots at -20 °C.
- dsRNA Synthesis
- Set up a reaction in a 0.2 ml PCR tube on ice and carry out an in vitro transcription reaction with T7 polymerase, following manufacturer's instructions, using ~ 500 ng template per reaction.
- Incubate the tube at 37 °C overnight.
- Digest the DNA template with DNase, following manufacturer's instructions.
- Heat the tube to 94 °C and hold at 94 °C for 3 min.
- To anneal the dsRNA, slowly cool the tube by 1 °C/min until it reaches 45 °C after 1 hr.
NOTE: Steps 3.3.2 through 3.3.5 can be performed in a thermocycler. - To ethanol precipitate of dsRNA, add 280 µl of RNase free water to 20 µl of reaction to adjust to a final volume of 300 µl. Add 30 µl of 3 M NaOAc (pH 5.2) and 650 µl of ethanol. Place tube in -20 °C freezer overnight.
- After centrifuging at 15,682 x g for 30 min at 4 °C, wash the dsRNA pellet with 70% ethanol. Dry pellet and dissolve in 20 µl injection buffer.
- Measure the concentration of the dsRNA using a spectrophotometer at A260. The ideal concentration is 3 – 5 µg/µl. Check the quality by running 5 µl of a 1:25 dilution of dsRNA on a 1% agarose gel at 90 V for 30 min. A single band of expected size will be readily visible.
- Store the dsRNA at -20 °C or colder.
NOTE: For every RNAi knockdown experiment, include a control dsRNA, which would not be expected to impact the process being studied. gfp dsRNA is an excellent control for most experiments. Prepare and inject the control dsRNA side-by-side with the experimental dsRNA.
4. Assembly of Microinjector and Micromanipulator
NOTE: See Figure 3.
- Connect nitrogen or other air supply to the pressure input of a pneumatic pump instrument. If a pneumatic pump is not available, apply pressure using a 50 ml syringe connected to fine tubing.
- Connect a foot switch to the remote connector of the pump to control the eject pressure flow.
- Attach a glass capillary holder to the eject pressure port of the pump with tube.
- Fix the capillary holder on a micromanipulator close to a dissecting microscope.
5. Embryonic RNAi
NOTE: Figure 4 shows a flowchart of D. maculatus embryonic RNAi.
- eRNAi Preparation
- Prepare an embryo collecting Petri dish lid (90 mm diameter). Place a piece of black filter paper to cover the inside of the lid. Put a standard microscope slide on the black paper.
- Dilute dsRNA to appropriate concentration with injection buffer. Use 2 – 3 µg/µl for initial experiments. Always keep dsRNA on ice prior to injection.
- Add food coloring at 1:40 dilution to the dsRNA and pipette gently several times to mix well.
- Collecting Embryos for Injection
NOTE: At 25 °C, nuclei in embryos 6 – 8 hr After Egg Laying (AEL) migrate towards the egg periphery53. A cellular blastoderm is established within 8 – 10 hr AEL53. To ensure embryos prior to cellular blastoderm stage are used for injection, 0 – 3 hr AEL embryos are recommended for use53. Embryo collection, preparation, and injection usually take less than 2 hr if the glass capillaries are in good shape. Therefore, the whole process can be completed within 5 hr AEL.- Collect embryos as described in steps 2.2-2.5.
- Tap the black construction paper to transfer embryos into collecting Petri dish lid.
- Stick double-sided tape along the edge of the slide.
- Using a paint brush, align the embryos on the tape perpendicular to the slide edge with the anterior or posterior end at the edge. Note that the anterior and posterior ends of early embryos are not readily distinguishable, thus on average half will be injected at the posterior end which is ideal.
- Loading the dsRNA into the Glass Capillary
- Take up 2 – 4 µl of dsRNA into a 20 µl microloader pipette tip.
- Insert the tip into a pre-pulled glass capillary (referred to as needle) and gently pipette the dsRNA solution into the capillary. Prepare needles from commercial glass capillaries using a micropipette puller. An example of an ideal pulled needle is shown in Figure 5. The length and taper of needles may need to be optimized after injection trials.
- Fix the glass capillary into the glass capillary holder.
- Assemble the capillary holder into the micromanipulator.
- Injecting dsRNA into Embryos
- Carefully transfer the slide with embryos onto the stage of the dissecting microscope.
- Move the slide to bring one embryo to the center of the field and focus microscope.
- Position the tip of the capillary with the micromanipulator while looking into the dissecting microscope. Bring the tip close to the end of the first embryo in the row.
- Switch on the nitrogen or other air supply.
- Set the solenoid input selector switch to vacuum.
- Adjust the eject pressure regulator to 10 – 15 psi. Adjust the pressure depending on the apparatus.
- Open the capillary if necessary. Once the tip is open, the colored dsRNA solution will fill the tip.
- Move the capillary tip forward to puncture the embryo. If the pressure setting is ideal, no embryonic fluid will flow into the capillary.
- Step on the foot switch to eject dsRNA solution into the embryo until an appropriate amount of solution is ejected. Figure 5 shows examples of embryo morphology after appropriate and inappropriate amounts of solution have been injected.
- Keep the capillary tip inside the embryo for ~ 2 sec, and then remove it.
- Move the capillary to the next embryo and repeat steps 5.4.8 – 5.4.10 until all embryos are injected. Change glass capillary if it gets clogged or breaks.
- Post-injection Recovery and Incubation
- After injection, place the slide in a Petri dish.
- Add a wet cotton ball to the Petri dish. Do not let the cotton ball touch the slide.
- Cover the Petri dish and wrap it with sealing film. Label the dish with the name of the dsRNA, concentration, date, time, and number of injected embryos.
- Place the Petri dish in a 30 °C incubator until the embryos hatch.
6. Parental RNAi
- Isolating Pupae for pRNAi
- Collect pupae from the colony.
- Sort male and female pupae under microscope (see Figure 6). Keep them in two separate Petri dishes in a 30 °C incubator to ensure that no mating occurs prior to injection.
- Check every day for eclosed adults. Pupation usually takes 5 – 7 days at 30 °C.
- Transfer eclosed males and females (see Figure 6) to two separate Petri dishes and feed them cat food every other day until they are ready for injection.
- Proceed to injection once there are enough females and males at appropriate age. Females 4 to 8 days post-eclosion are best. For a typical analysis, 8 – 12 females are used. An equal number of males are needed for mating54.
- Injecting dsRNA into Female Abdomen
- Prepare dsRNA on ice (5.1.2 and 5.1.3).
- Attach a 32-gauge needle to a 10 µl syringe.
- Anaesthetize females on CO2 stage.
- Load dsRNA solution into the syringe without taking up any air.
- Hold a female ventral side up with one hand and hold the syringe with the other.
- Gently penetrate the segmacoria (membrane) between sternites 2 and 3 with the tip of the needle. Figure 7 shows a female during injection. If the needle does not penetrate the tissue easily, angle the needle upward and slowly press down. Insert approximately 2 mm of the needle into the body.
- Check the syringe scale while slowly pushing the plunger, injecting ~ 2 µl per female.
- After injecting, hold the needle still for at least 5 sec before removing it from the female's abdomen.
- Transfer the injected females to a Petri dish (90 mm diameter) and feed with cat food.
- Label the Petri dish with the dsRNA name, amount injected, date, and number of females.
- Post-injection Recovery and Mating
- Incubate the Petri dish in a 30 °C incubator.
- After 24 hr, transfer injected females to a mini cage.
- Add to the cage an equal number of uninjected young males, and label the cage.
- Add a weighing boat with cat food into the cage.
- Leave the cage in a 30 °C incubator to allow mating. After 24 hr, females should start to lay eggs and embryos can be collected daily or at required time intervals.
7. Phenotypic Analysis after RNAi
NOTE: At 30 °C, it takes ~ 55 hr for eggs to hatch50.
- eRNAi Viability Analysis
- Calculate hatch rate by comparing the number of hatched larvae to the total number of injected embryos. Be sure to include a negative control injection as embryos may be harmed or killed by the injection procedure. Typical hatch rates vary from 30% – 60%. Beware of hatched larvae eating unhatched eggs.
- pRNAi Viability Analysis
NOTE: If female viability is impacted by the gene targeted by RNAi, females injected with specific dsRNA but not negative control dsRNA will begin to die. If egg production or egg laying is impacted, females will exhibit partial or complete sterility. Score female survival compared to negative controls or compare total number of eggs laid by experimental and control females (7.2.3).- Set up embryo collection following step 2.2.
- After 24 hr, collect embryos following steps 2.4 – 2.5.
- Count the total number of the embryos.
- Transfer the embryos to a 90 mm Petri dish. Label the Petri dish with the gene targeted, collection date, and number of embryos.
- Incubate the embryos in a 30 °C incubator until they hatch. Since hatched larvae feed on unhatched embryos, check the Petri dish frequently and separate hatched larvae from unhatched embryos using forceps (see Discussion).
- Count the number of hatched embryos and calculate the hatch rate.
- Examining Cuticle Defects in Hatched Larvae
- Collect hatched larvae in a 1.5 ml microcentrifuge tube.
- In a fume hood, add 1 ml of Fixation Solution.
- Leave the microcentrifuge tube at 4 °C at least overnight to let the solution penetrate the larval tissue.
- To visualize, remove as much fixative from the tube as possible.
- Rinse larvae at least three times with PBST.
- Transfer larvae to a multi-well glass plate with PBST using a P-1000 pipette tip with the end cut off to widen it.
- Under a dissecting microscope, stretch larvae using forceps to examine defects. It is critical to stretch out larvae to examine defects as their bodies contract in fixative.
- Examining Cuticle Defects in Unhatched Larvae
Note: This procedure is useful for examination of early embryonic phenotypes, especially for embryos that do not survive to hatching.- For pRNAi, add PBST into the Petri dish (7.2.4) and transfer unhatched embryos to a 1.5 ml microcentrifuge tube using a P-1000 pipette tip with the end cut off. For eRNAi, add PBST into the Petri dish (5.5.4), brush unhatched embryos off the microscope slide and transfer them to a microcentrifuge tube.
- Fix the embryos with Fixation Solution and rinse them with PBST for visualization following steps 7.3.2 – 7.3.6.
- Using forceps, dissect embryos out of the eggshell under a dissecting microscope in PBST.
- Transfer the embryos back to 1.5 ml microcentrifuge tube (~ 200 µl of embryos in each tube).
- Remove as much solution as possible.
- Add 1 ml of 90% lactic acid/10% EtOH. Leave the centrifuge tube in a 60 °C incubator at least overnight.
NOTE: Embryos can be left at 60 °C for several days. - To mount the embryos, pipette them onto a microscope slide using a P-1000 pipette tip with the end cut off.
- Manually position the embryos with forceps to an ideal orientation.
- Cover the embryos with a cover-slip and visualize under microscope using DIC.
- Examining Cellular and Molecular Defects
NOTE: This procedure is useful for investigating the underlying causes of cuticle phenotypes or lethality that is not accompanied by cuticle defects.- For pRNAi, separate embryos from cotton balls at appropriate development stage and transfer them into an embryo collecting basket. The collection basket can be the same or similar to that used for Drosophila embryo collections.
- Make the egg basket from a 25 ml plastic scintillation vial with the bottom of the vial removed, and a circle cut out of the lid (roughly 1 cm diameter). Place a circle of super fine mesh about 2.5 cm in diameter inside the lid, and then screw the scintillation vial on and use upside-down.
- As the mesh can be easily removed once the embryos are washed, immerse the mesh in a microcentrifuge tube with water to recover any embryos sticking to the mesh.
- For eRNAi, at appropriate stage, add PBST to the Petri dish. Brush the embryos off the microscope slide and transfer them to an embryo collecting basket.
NOTE: Fixation, in situ hybridization, antibody staining, and nuclear staining protocols can be found in Xiang et al. 201553.
- For pRNAi, separate embryos from cotton balls at appropriate development stage and transfer them into an embryo collecting basket. The collection basket can be the same or similar to that used for Drosophila embryo collections.
The authors' lab has used RNAi technology to study the functional evolution of genes regulating segmentation in insects53,55. While all insects are segmented, the genes regulating this process appear to have diverged during insect radiations26,38,56-63. Genetic screens in Drosophila identified a set of nine pair-rule segmentation genes that are responsible for promoting the formation of body segments64-70. Here, the ortholog of one of these genes, paired (prd), is used to document the utility of RNAi for studying gene function in D. maculatus.
eRNAi and pRNAi were each effective in demonstrating roles for Dmac–prd in segment formation in this species. 2 – 3 µg/µl of dsRNA designed to target Dmac–prd (Figures 8B, 8D and 8F) was compared to gfp dsRNA, injected as negative control (Figures 8A, 8C and 8E).
Control offspring hatched with one pigmented stripe per segment. Neighboring pigmented stripes were separated by a non-pigmented gap (Figure 8A). After prd pRNAi, affected offspring hatched with fused neighboring pigmented stripes, indicating segmental boundaries were defective (Figure 8B). Depending on phenotypic severity, one or several fusions were detected in affected larvae. Nevertheless, fusions consistently appeared at the boundary regions between T2/T3, A1/A2, A3/A4, A5/A6, A7/A8, indicating pair-rule-like defects. Cuticle phenotypes after prd knockdown showed loss of abdominal segments as well as shortened body length (Figure 8D). Engrailed (En) antibody staining was performed to examine molecular defects in early embryos after prd knockdown. While control embryos showed striped En expression with equal intensity in every segment (Figure 8E), reduced En expression was detected in alternate stripes in offspring from Dmac-prd dsRNA injected females (asterisks in Figure 8F). The pattern of reduced En expression is consistent with the defective cuticle pattern observed in affected hatched larvae. For more detailed results of phenotypes after prd knockdown in D. maculatus, see Xiang et al. 201553.

Figure 3: Injection Apparatus. Photograph of the dissection microscope and micromanipulator used to inject D. maculatus embryos is shown. Nitrogen supply is connected to the pressure input port of a pneumatic pump. Glass capillary holder is connected to the eject pressure port of the pump. Foot switch is connected to the pump. Please click here to view a larger version of this figure.
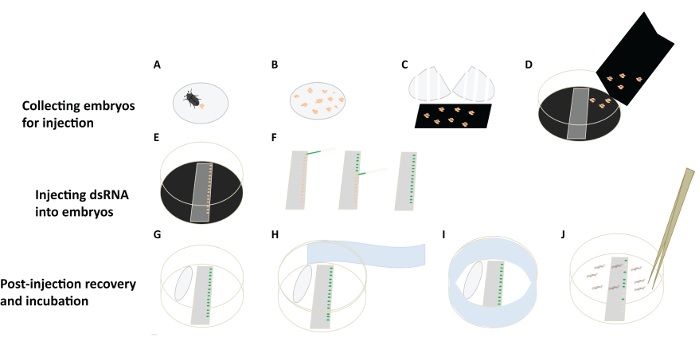
Figure 4: A Flowchart for D. maculatus Embryonic RNAi. (A-D) Collecting embryos for injection. Females lay embryos (orange) in cotton balls (grey circle). Separate embryos from cotton balls and let them drop onto a piece of black construction paper. White bars indicate tears in cotton ball. Transfer embryos to a collecting Petri dish lid, lined with black paper. (E, F) Injecting dsRNA into embryos. Align embryos on slides on double stick tape and inject them individually under a dissecting microscope. Needle with green food dye is shown. (G-J) Post-injection recovery and incubation. Place a wet cotton ball in the Petri dish to provide humidity. Cover the Petri dish and wrap it with sealing film (light blue). Place the Petri dish in an incubator and remove hatched larvae for phenotypic analysis. Please click here to view a larger version of this figure.
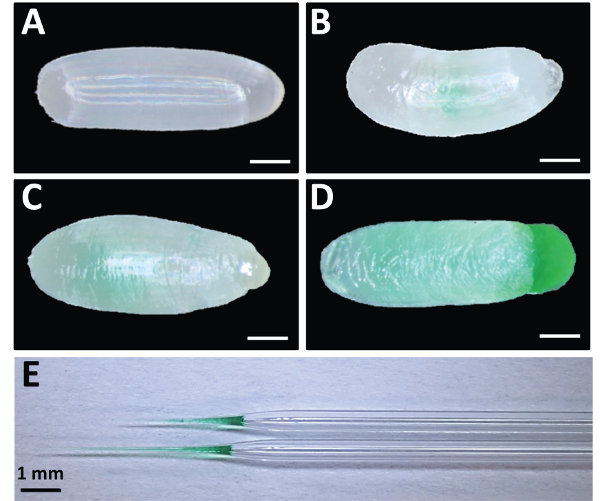
Figure 5: Good vs. Bad Embryonic Injection Examples. (A) Uninjected embryo. (B) Embryo injected with too little dsRNA. (C) Embryo injected with appropriate amount of dsRNA. (D) Broken embryo with overflowing dsRNA caused by over-injection. Food coloring was added to dsRNA for visualization. (E) Examples of pulled capillary tubes used as needles for injection. Note the taper and tip length for two examples of functional needles. Scale bars for A-D represent 200 μm. Please click here to view a larger version of this figure.
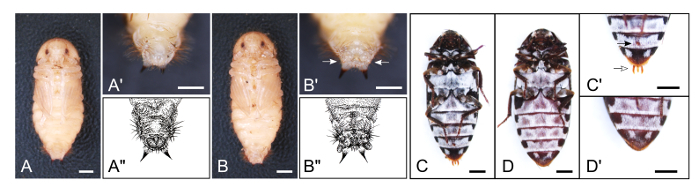
Figure 6: Male and Female Adults and Pupae. (A) Ventral view of a male pupa. (A') Magnification of posterior part of A. (A") Illustration of male pupa genitalia. (B) Ventral view of a female pupa. (B') Magnification of posterior part of B. Female pupae have two genital papillae (white arrows). (B") Illustration of female pupa genitalia. (C) Ventral view of a male adult. (C') Magnified view of posterior part of C. Note that male adults have trident-like genitalia and a circular lobe-like structure on the 4th sternite (white and black arrows, respectively). (D) Ventral view of a female adult. (D') Magnified view of posterior part of D. For all panels, scale bars indicate 1 mm. For details, see Xiang et al. 201553. Please click here to view a larger version of this figure.

Figure 7: A Female during Injection. Females are anesthetized and placed ventral side up. A needle penetrating the segmacoria between the 2nd and 3rd sternites to inject dsRNA is shown. Please click here to view a larger version of this figure.
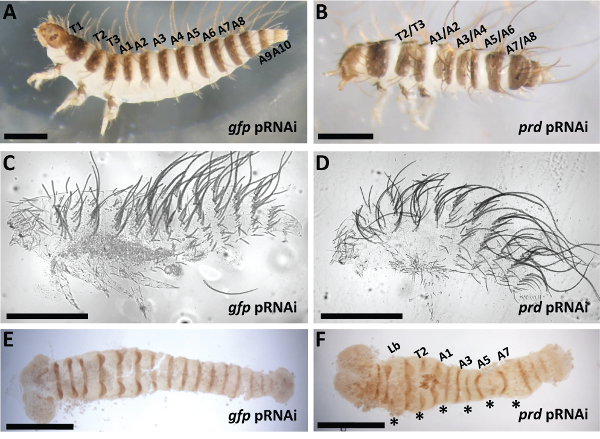
Figure 8: Representative Phenotypes after Dmac-prd pRNAi. (A) Control injection. Lateral view of a hatched wild type-like gfp pRNAi offspring with three thoracic segments and ten abdominal segments. Each segment has setae and pigmented stripes. (B) Lateral view of a hatched prd pRNAi offspring. The gap between labeled neighboring pigmented stripes is narrowed or completely missing. (C) Cuticle phenotype of an unhatched control wild type-like embryo. (D) Cuticle phenotype of an unhatched prd pRNAi offspring with fewer abdominal segments. (E,F) Dissected germband from: (E) Control, gfp pRNAi has evenly expressed En in every segment, (F) prd pRNAi, En expression is reduced in alternate segments (asterisks). For all panels, scale bars indicate 500 μm. Please click here to view a larger version of this figure.
