A schematic workflow of the protocol is provided in Figure 1.
ECM extraction protocol
The efficiency of the extraction can be monitored by running aliquots form each extract on Bis-Tris acrylamide gels and using silver staining for visualization. Figure 2A shows the complementarity of the NaCl, SDS and GuHCl extracts after sequential extraction. This QC allows for identification of potential issues with sample quality such as excessive protein degradation. After extraction, ECM glycoproteins are abundant in the GuHCl extracts (Figure 2B).
Deglycosylation
To assess the efficiency of deglycosylation, a non-deglycosylated control should be run in parallel. Deglycosylation times have to be suitable to achieve a complete and homogeneous removal of sugar residues, as exemplified in Figure 3A. Figure 3B shows a representative example of samples efficiently deglycosylated by the addition of enzymes for GAG removal and deglycosylation enzymes that target smaller N- and O-linked oligosaccharides.
Glycoproteomics
The protocol for assessment of the occupancy of NxT/S sequons improves protein sequence coverage for ECM glycoproteins after MS (Figure 3C) and allows for an initial screening of the presence of glycoproteins. This helps to reduce the search time for glycopeptides, as databases can be customized to contain previously identified glycoproteins. HCD-ETD fragmentation is used for identification and compositional characterization of oligosaccharides attached to ECM glycoproteins. Figure 4A displays a representative spectrum obtained for a peptide labeled with 18O after deglycosylation (indirect glycopeptide analysis). Figure 4B is a representative example of a spectrum obtained after analysis of intact glycopeptides from ECM extracts (direct glycopeptide analysis).
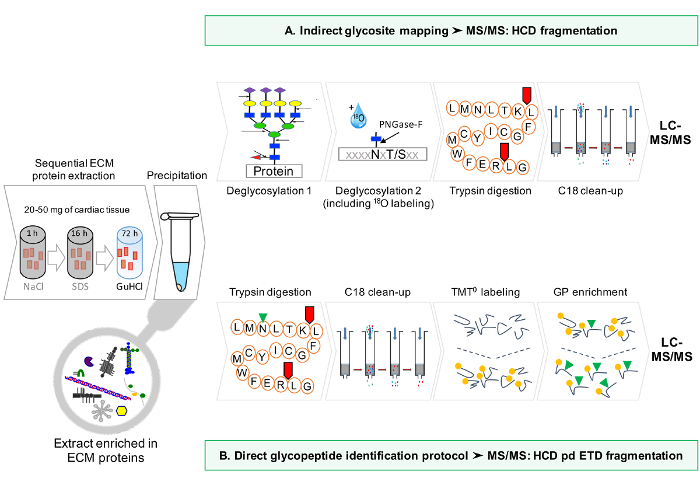
Figure 1: Method Overview. (A) After sequential enrichment for ECM proteins, LC-MS/MS analyses are performed on the deglycosylated extracts. (B) Alternatively, non deglycosylated ECM extracts are further enriched for glycopeptides. Please click here to view a larger version of this figure.
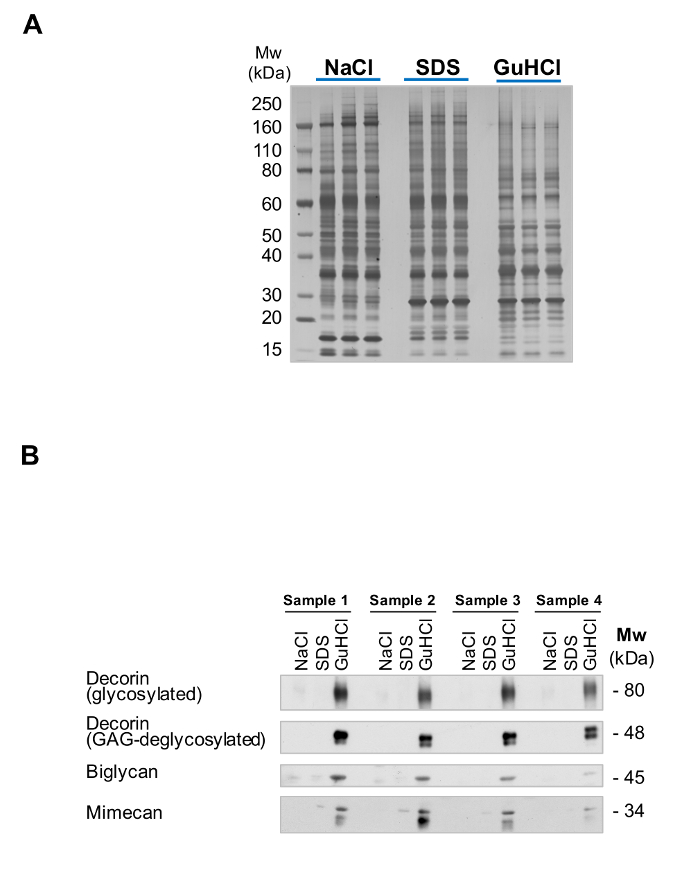
Figure 2: Extraction of ECM Proteins. (A) The 3 different extracts from the sequential extraction procedure ("English Quickstep") are complementary in their protein content. While SDS extracts are enriched in intracellular proteins, GuHCl extracts contain the majority of ECM proteins. Successful fractionation is visualized by the different silver staining pattern. (B) ECM proteins such as the small leucine-rich proteoglycans decorin, biglycan and mimecan are predominantly detected in the GuHCl extracts, with little presence in the SDS and NaCl extracts. Please click here to view a larger version of this figure.
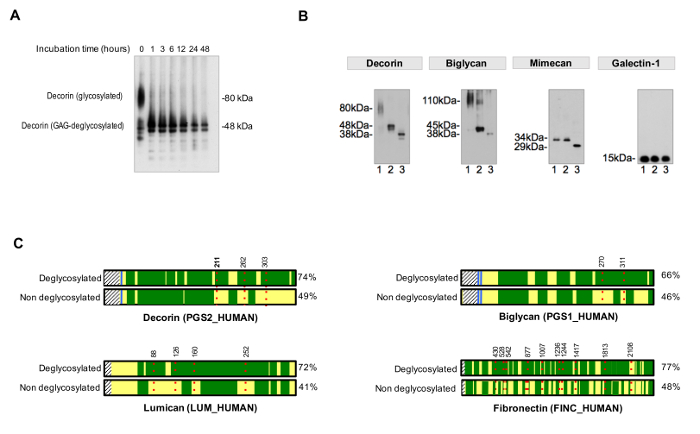
Figure 3. Analysis of Glycosylation. (A) Appropriate incubation times are required for complete deglycosylation. The example shows the effect of incubation time during removal of glycosaminoglycan chains from the glycoprotein decorin. (B) ECM glycoproteins are decorated with large and repetitive glycosaminoglycan chains and short and diverse N- and O-linked oligosaccharides. Lane 1 on each of the immunoblots represents untreated cardiac extracts. Lane 2 contains extracts treated with enzymes that digest glycosaminoglycans. Samples in lane 3 contain, in addition, enzymes for the removal of N- and O-linked oligossacharides. Galectin-1 is not glycosylated, hence there is no shift in protein size. Adapted from Lynch M, et al.4 (C) In LC-MS/MS analysis, samples treated with PNGase-F in the presence of H218O achieve better sequence coverage (%, on the right side) compared to non-deglycosylated samples. Dark green areas represent sequence coverage by LC-MS/MS. The red, dotted lines represent glycosites, with numbers indicating their amino acid position. Detection of glycosylation of decorin at position Asn211 (N, highlighted in bold) is shown in detail as an example in Figure 4. Please click here to view a larger version of this figure.
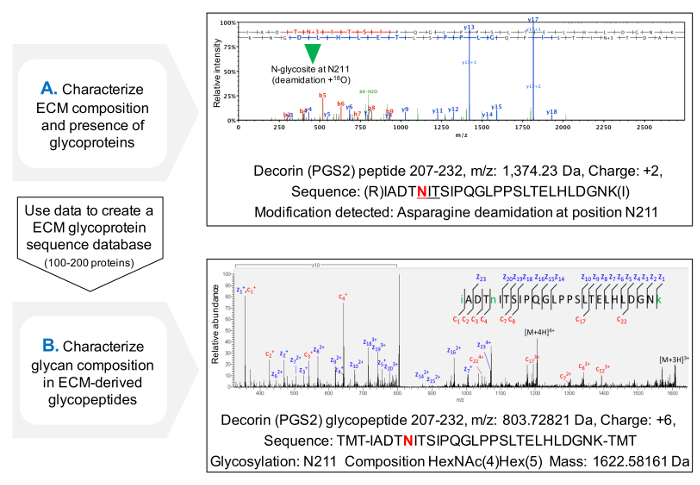
Figure 4. Glycopeptide Analysis by MS. (A) Using a shotgun proteomics approach on ECM enriched extracts, glycopeptides can be identified by the presence of deamidated asparagines within NxT/S sequons and labeled with 18O. The example shows a HCD MS/MS spectrum for a peptide of decorin containing the previously glycosylated Asn211. The data obtained can be used to create a customized database of ECM glycoproteins. (B) HCD-ETD fragmentation is used to analyze the glycopeptide enriched ECM extract. The ETD MS/MS spectrum allows the characterization of glycan composition. Please click here to view a larger version of this figure.
| A. Stock solutions | |
| DTT (Dithiotreitol, C4H10O2S2) | 100 mM DTT in ddH2O.1 |
| EDTA (Ethylenediaminetetraacetic acid, C10H16N2O8) | 250 mM EDTA in ddH2O, pH 8.0. |
| GuHCl (Guanidine hydrochloride, CH6ClN3) | 8 M GuHCl in ddH2O. |
| IAA (Iodoacetamide, C2H4INO) | 500 mM IAA in ddH2O.1,2 |
| Na acetate (Sodium acetate, C2H3NaO2) | 1 M Na acetate in ddH2O, pH 5.8. |
| NaCl (Sodium chloride, NaCl) | 1 M NaCl in ddH2O. |
| Na phosphate dibasic (Disodium phosphate, Na2H2PO4) | 1 M Na phosphate dibasic in ddH2O, pH 6.8. |
| SDS (Sodium dodecyl sulfate, NaC12H25SO4) | 1% SDS (35 mM) in ddH2O.3 |
| TFA (Trifluoroacetic acid, C2HF3O2) | 10% TFA (1.2 M) in ddH2O. |
| TEAB (Triethylammonium bicarbonate, C7H17NO3) | 1M TEAB in in ddH2O, pH 8.5 |
| Thiourea (Thiourea, CH4N2S) | 3 M thiourea in ddH2O. |
| Tris-HCl (Tris-hydrochloride (NH11C4O3[HCl]) | 100 mM Tris–HCl in ddH2O, pH 7.5. |
| Urea (Urea, CH4N2O) | 9 M urea in ddH2O. |
| B. Reaction buffers | |
| C18 clean-up equilibration buffer | 1% ACN, 0.1% TFA in ddH2O |
| C18 clean-up column wash buffer | 80% ACN, 0.1% TFA in H2O |
| C18 clean-up elution buffer | 50% acetonitrile, 0.1% TFA in ddH2O |
| Deglycosylation Buffer (4x) | 600 mM NaCl and 200 mM Na phosphate in ddH2O, pH 6.8. |
| GuHCl buffer4 | 4 M guanidine hydrochloride, 50 mM Na acetate and 25 mM EDTA in ddH2O, pH 5.8. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| NaCl buffer4 | 0.5 M NaCl, 10 mM Tris-HCl and 25 mM EDTA in ddH2O, pH 7.5. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| PBS (1x) | 1.7 mM KH2PO4, 5 mM Na2HPO4, 150 mM NaCl, pH 7.4. Add 25 mM EDTA and 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| Sample buffer (4x) | 100 mM Tris, 2% SDS, 40% Glycerol, 0.02% bromophenol blue in ddH2O, pH 6.8. Add 10% ß-mercaptoethanol before use. |
| SDS buffer4 | 0.1 % SDS and 25 mM EDTA in ddH2O. Add 1:100 (v:v) of cocktail of proteinase inhibitors before use. |
| C. Enzymes | |
| Chondroitinase ABC5 | 0.5 U/mL in deglycosylation buffer (1x) |
| Keratanase5 | 0.1 U/mL in deglycosylation buffer (1x) |
| Heparinase II5 | 0.1 U/mL in deglycosylation buffer (1x) |
| α2-3,6,8,9-Neuraminidase (sialidase)5 | 0.025 U/mL in deglycosylation buffer (1x) |
| β1,4-Galactosidase5 | 0.015 U/mL in deglycosylation buffer (1x) |
| β-N-Acetylglucosaminidase5 | 0.25 U/mL in deglycosylation buffer (1x) |
| Endo-α-N-acetylgalactosaminidase (O-glycosidase)5 | 0.013 U/mL in deglycosylation buffer (1x) |
| PNGase-F(N-glycosidase-F)6 | 50 U/mL in H218O |
| Trypsin | 0.01 µg/µL in TEAB buffer |
| Table NOTES. | |
| 1 Keep stock solution frozen at -20 °C. | |
| 2 IAA should be kept protected from light. | |
| 3 SDS readily crystallizes at < 20 °C. In order to facilitate solubilization of 1% SDS (stock solution), warm the buffer under hot tap water. | |
| 4 Extraction buffers can be stored at RT. Add broad-spectrum cocktail of proteinase inhibitors as indicated before use. | |
| 5 These enzymes should be added during the first deglycosylation step. | |
| 6 PNGase-F should be only added during the second deglycosylation step. | |
Table 1: Stock Solutions, Reaction Buffers and Enzymes. This table lists the composition of each stock solution and reaction buffer required for the extraction and subsequent processing (including enzymatic digestions) of cardiac ECM proteins prior to MS analysis.
