In order to confirm the presence of the correct number of gRNA inserts in the concatemer vector, restriction digestion is performed with enzymes (EcoRI + BglII) flanking all gRNA expressing cassettes (each cassette size is ~400 bp, Figure 1). For example, when generating a 4 gRNA- concatemer vector, the expected size of the lower band in the agarose gel is approximately 1.6 Kbp; any band lower than this indicates that not all of the 4 gRNA cassettes are inserted into the vector (Figure 2A). In addition, it is always recommended to check that all BbsI recognition sites are lost and the enzyme does not cut the vector (Figure 2B).
Once the constructs have been confirmed, they can be delivered to mouse intestinal organoids by electroporation to achieve optimal levels of transfection efficiency (up to 70%), as shown by the GFP control (Figure 3).
Finally, to functionally test the efficiency of this strategy, intestinal organoids transfected with Cas9 and concatemer vectors against Axin1/2 and Rnf43/Znrf3 were cultured in EN (R-spondin withdrawal) and EN + IWP2 (R-spondin and Wnt withdrawal, IWP2: Porcupine inhibitor, 2.5 μM) media for a minimum of 3 passages (Figure 4). While untransfected WT organoids died under both conditions, Axin1/2 knockout organoids survived in both due to downstream activation of the Wnt pathway; in addition, Rnf43/Znrf3 mutant organoids survive in the absence of R-spondin but cannot survive in the presence of IWP2, which causes depletion of the Wnt that activates the pathway. Taken together, these observations demonstrate that knockout of these pairs of paralogues is possible by generating the expected organoid phenotype. Details of these results have been published in Developmental Biology5.
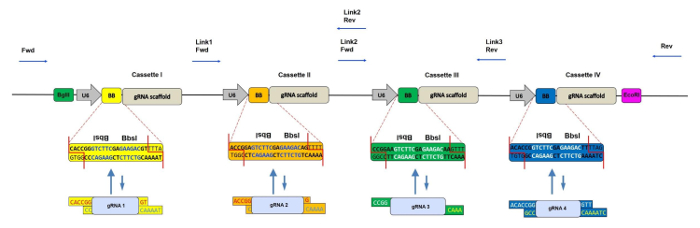
Figure 1: Schematic Representation of the CRISPR-concatemer with 4 Cassettes. Scheme of the 4 gRNA-concatemer vector with each 400 bp cassette containing a U6 promoter, two inverted repeated BbsI sites (also indicated as BB) and gRNA scaffold in this order. During the shuffling reaction, BbsI sites are replaced by gRNA fragments with matching overhangs and consequently lost. Binding sites of the sequencing primers for checking the correct insertion of gRNA oligos are shown by the blue arrows. Fwd = forward primer, Rev = reverse primer, Link 1/2/3 = linker regions 1/2/3. Please click here to view a larger version of this figure.
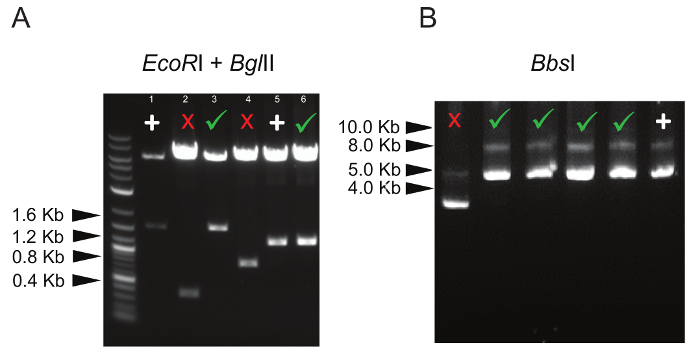
Figure 2: Representative Digestion Patterns of Concatemer Vectors. (A) Double digestion of 3 and 4 gRNA-concatemer vectors with EcoRI and BglII. The correct digestion pattern is marked by a green tick, whereas vectors with only 1 or 2 gRNA insertions are marked by a red cross. Lane 1 shows digestion of a 4 gRNA-concatemer parental vector used as positive control (marked by "+"); similarly, lane 5 shows digestion of a 3 gRNA-concatemer parental vector, marked by "+". (B) Digestion with BbsI, showing the correct size of undigested concatemer vectors (indicated by the green ticks). Digestion of a gRNA-containing concatemer vector that has lost BbsI sites is used as a positive control and is marked by "+". Please click here to view a larger version of this figure.
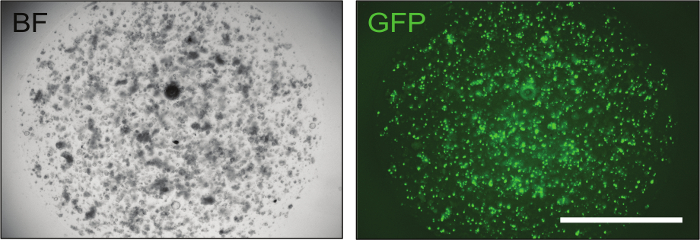
Figure 3: Representative Image of Successfully Electroporated Intestinal Organoids. Transfection of a GFP plasmid is instrumental to evaluate transfection efficiency. Approximately 24 h after electroporation, organoids containing a small number of cells are already visible and, if the electroporation procedure was successful, up to 70% of them displays green fluorescence. BF = bright field, GFP = green fluorescent protein. Scale bar = 2,000 µm. Please click here to view a larger version of this figure.
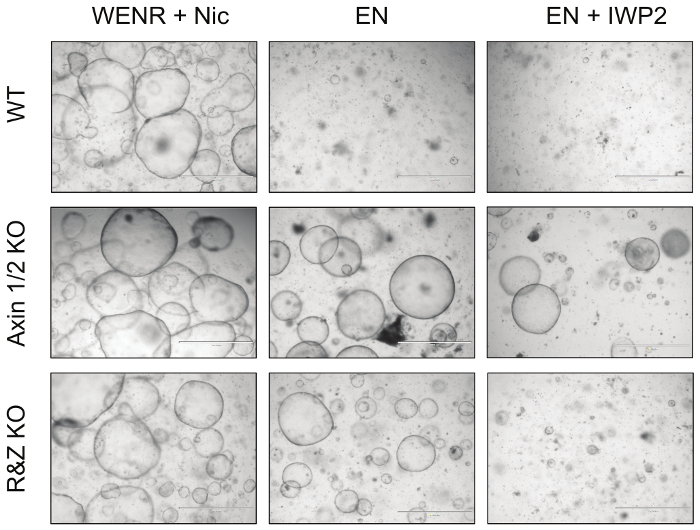
Figure 4: Representative Images of Mutant Intestinal Organoids. Knockout of the negative regulators of the Wnt pathway Axin1 and Rnf43, together with their paralogues, renders the intestinal organoids resistant to growth factor deprivation. In particular, Axin1/2 knockout organoids (Axin1/2 KO) can grow in the absence of both R-spondin (EN: EGF + Noggin) and Wnt (EN + IWP2: EN + Porcupine inhibitor), whereas Rnf43/Znrf3 mutant organoids (R&Z KO) can only survive in the absence of R-spondin (EN). In contrast, WT organoids can only survive in the control culture condition, WENR + Nic (Wnt + EGF + Noggin + R-spondin + Nicotinamide). Scale bars = 1,000 µm. Please click here to view a larger version of this figure.
| Basal medium | Comments | |
| Store at 4 °C for 4 weeks | ||
| Cell culture medium | 500 mL | See table of materials |
| L-Glutamine 100x | 5 mL | |
| Buffering agent 1 M | 5 mL | See table of materials |
| Penicillin Streptomycin 100x | 5 mL | |
| WENR + Nic (Wnt + EGF + Noggin + R-spondin + Nicotinamide) | ||
| Store at 4 °C for 2 weeks | ||
| Basal medium | up to 50 mL | |
| Neuronal cell serum-free supplement (50x) | 1 mL | See table of materials |
| Neuronal cell serum-free supplement (100x) | 500 μL | See table of materials |
| n-Acetylcysteine (500 mM) | 125 μL | |
| mouse EGF (100 μg/mL) | 25 μL | |
| mouse Noggin (100 μg/mL) | 50 μL | |
| R-Spondin conditioned medium | 5 mL | |
| Wnt3a conditioned medium | 25 mL | |
| Nicotinamide (1 M) | 250 μL | |
| EN + CHIR + Y-27632 (EGF + Noggin + CHIR + Y-27632) | ||
| Store at 4 °C for 2 weeks | ||
| Basal medium w/o Penicillin Streptomycin | up to 20 mL | |
| Neuronal cell serum-free supplement (50x) | 400 μL | See table of materials |
| Neuronal cell serum-free supplement (100x) | 200 μL | See table of materials |
| n-Acetylcysteine (500 mM) | 50 μL | |
| mouse EGF (100 μg/mL) | 10 μL | |
| mouse Noggin (100 μg/mL) | 20 μL | |
| Y-27632 (10 μM) | 20 μL | |
| CHIR99021 (8 μM) | 10 μL | |
| EN (EGF + Noggin) | ||
| Store at 4 °C for 4 weeks | ||
| Basal medium | up to 50 mL | |
| Neuronal cell serum-free supplement (50x) | 1 mL | See Table of materials |
| Neuronal cell serum-free supplement (100x) | 500 μL | See Table of materials |
| n-Acetylcysteine (500 mM) | 125 μL | |
| mouse EGF (100 μg/mL) | 25 μL | |
| mouse Noggin (100 μg/mL) | 50 μL |
Table 2: Organoid Media Composition.
