As representative examples of images obtained from the protocol described here, mouse brain sections were stained with antibodies recognizing different components of the NVU including the basement membrane, astrocytes, pericytes, and endothelial cells (see Table of Materials for specific antibodies used) (Figure 1A, D, and E). At this resolution it is possible to distinguish the individual astrocytic processes and end-feet that are in direct contact with capillaries.
To highlight the suitability of this protocol for detecting intracellular structures, brain sections from animals peripherally injected with the human anti-Tau mAb86 antibody16 were stained with a fluorescently labelled anti-human antibody (Figure 1B). mAb86 is known to specifically target neurons expressing a pathological form of Tau16. Using the protocol described herein, mAb86 was detected with diffraction-limited resolution within individual vesicular structures within neurons (Figure 1B-C). In addition, endogenous mouse IgG was detected in intracellular structures within endothelial cells but not in pericytes (Figure 1D-E and Figure 2).
The acquisition of high-resolution confocal z-stacks of the brain vasculature allows for three-dimensional segmentation of capillaries and intracellular vesicles at the BBB. Figure 2 shows an example of the process of rendering and segmenting a capillary labelled with CollagenIV and mouse IgG-positive intracellular vesicles. By quantifying a full dataset of segmented images, for example by measuring the number of vesicles per capillary volume, it is possible to study changes in intracellular transport processes under different conditions. Figure 3B-C shows the differences in mIgG vesicle number and fluorescence intensity corresponding to mIgG at the brain parenchyma, respectively, upon pericyte depletion in the pdgf-bret/ret mouse model as previously reported17. The same approach was also recently used to analyze changes in intracellular transport at the BBB between different brain regions18.
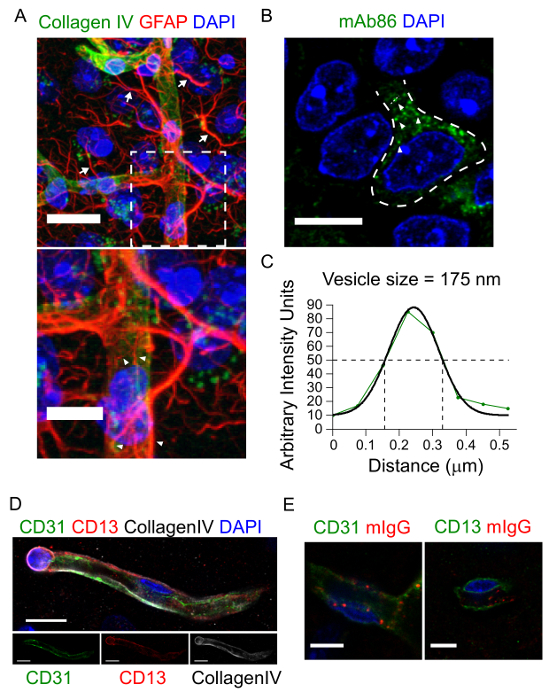
Figure 1: Labelling of multiple cell types and subcellular structures of the neurovascular unit. Representative images of the neurovascular unit (A and D) and intracellular vesicles contained within neurons (B) or endothelial cells (E) obtained with this protocol. The maximum intensity projection image in A (top) shows the distribution of GFAP-positive astrocytes (red) surrounding capillaries labelled by CollagenIV (green). Arrows point to individual astrocytic processes. Scale bar = 20 µm. At this resolution, the individual astrocyte processes and end-feet are clearly visible, as shown in the zoomed image of the boxed region (bottom). Arrowheads point to astrocytic end-feet. Scale bar = 10 µm. The image in B shows the accumulation of a peripherally-injected antibody, mAb86 (green), within a hippocampal neuron. Arrowheads point to individual mAb86-positive vesicles. Scale bar = 10 µm. The graph in C shows the line profile intensity of a single vesicle. The vesicle size was estimated from the full width at half maximum of a Gaussian fit (black solid line) of the intensity curve (green line and circles). The images in D show a three-dimensional reconstruction of an endothelial cell (green) surrounded by a pericyte (red) within the basal lamina (CollagenIV, grey). The lower panels show the individual fluorescence channels. Scale bar = 10 µm. The images in E show the localization of mIgG (red) in intracellular vesicles within endothelial cells (left panel, CD31 in green) but not in pericytes (right panel, CD13 in green). In all images, DAPI stained nuclei are shown in blue. Scale bar = 5 µm. Panels D and E have been modified from reference17. Please click here to view a larger version of this figure.
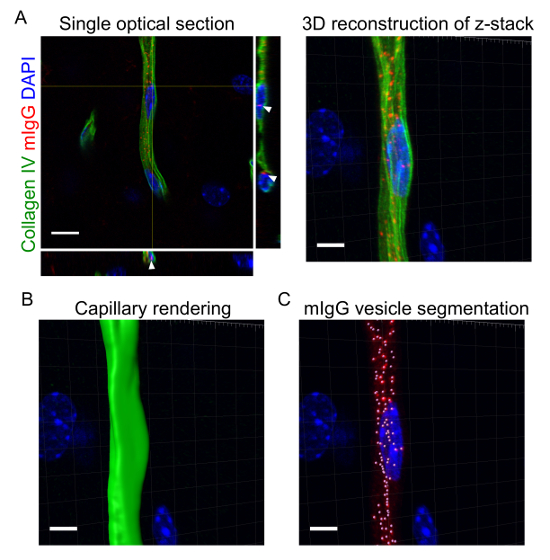
Figure 2: Three-dimensional rendering of capillaries and intracellular vesicles at the blood-brain barrier. With the protocol described, high-resolution confocal images of CollagenIV-positive capillaries (green) and mouse IgG intracellular vesicles (red) were acquired (A). The left panel shows a single optical section with cross-sections. The arrows point to individual mIgG-positive vesicles within brain endothelial cells. The panel on the right shows the 3D reconstruction of the full z-stack using image processing software (see Table of Materials). The capillary volume (B) and individual vesicles (C) were rendered in three dimensions and quantified using image processing software (see Table of Materials). In all images, DAPI stained nuclei are shown in blue. Scale bars = 5 µm. Please click here to view a larger version of this figure.
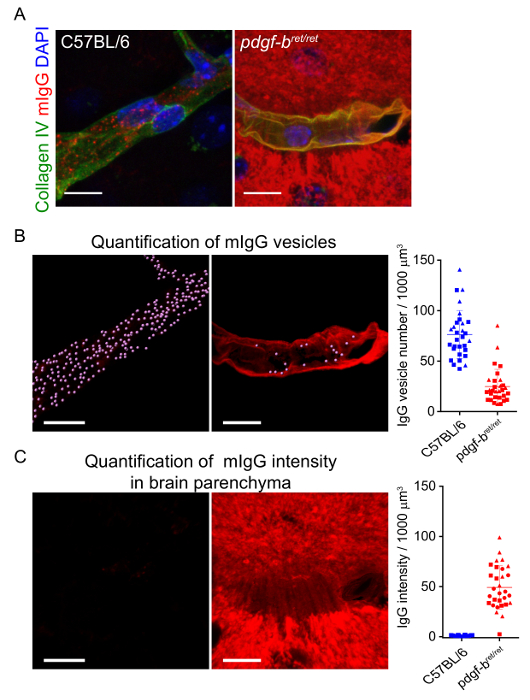
Figure 3: Quantification of mIgG intracellular localization at the BBB. Representative images showing (A) three-dimensional reconstructions of capillaries (labelled with collagenIV, green) and the distribution of mIgG (red) in C57BL/6 mice and in pdgf-bret/ret pericyte depleted mice previously described in19. Scale bars = 10 µm. The images in B show the segmentation of intracellular vesicles within the CollagenIV mask. The graph in B shows the quantification and comparison of mIgG vesicle number per volume of capillary. Each point represents measurements from individual capillary segments. The solid line shows the mean and the error bars represent the standard deviation of the data. The images in C show the mIgG fluorescence signal outside the CollagenIV mask. Similarly, the graph in C shows the quantification and comparison of mIgG fluorescence intensity in the brain parenchyma between C57BL/6 mice and pdgf-bret/ret pericyte depleted mice. Fluorescence intensity units were normalized by the average mIgG parenchyma intensity measured in all C57BL/6 mice. This figure has been modified from reference17. Please click here to view a larger version of this figure.
