1. Coupling Gal4- and QF-mediated Htt Transgene Expression in Drosophila
- Collect and/or generate transgenic Drosophila melanogaster lines containing tissue-specific Gal4 or QF "drivers," as well as lines containing wild-type or mutant Htt transgenes downstream of the Gal4-UAS38 or QF-QUAS39. Ensure that the proteins that are expressed from these transgenes are fused to fluorescent proteins or epitope-tagged to allow for differentiation of mutant and wild-type Htt transgene products in the same fly. See Figure 1.
NOTE: We typically use exon 1 fragments of the human Htt gene27. However, transgenic flies can instead be generated to express longer Htt fragments or other genes if desired. - Plan the genetic strategy such that mutant and wild-type Htt transgenes will be expressed in distinct, non-overlapping cell populations.
- If any expression overlap occurs, mutant and wild-type Htt proteins synthesized in the same cells will co-aggregate and prevent detection of prion-like events. To avoid this, use the Gal4 and QF specific repressors, Gal80 and QS40, respectively (Figure 1). Selecting developmentally-controlled Gal4 and/or QF drivers can help to restrict expression of mutant Htt to post-mitotic cells, eliminating the possibility that cell division might contribute to cell-to-cell aggregate spreading.
- Using standard culturing conditions, mate the flies to generate progeny that express mutant Htt via QF (or Gal4) in a "donor" cell population and wild-type Htt via Gal4 (or QF) in a "recipient" cell population. See Figure 1 for a schematic showing a possible genetic combination.
NOTE: The QF and Gal4 drivers that were used to generate the data shown in Figures 1 – 4 and in our previous publication27 include the DA1 olfactory receptor neuron (ORN) driver, Or67d-QF, and the pan-glial driver, repo-Gal4. - Generate control flies in parallel that express wild-type Htt in both QF- and Gal4-labeled cell populations.
- Collect progeny of the desired genotype, and age the animals as appropriate.
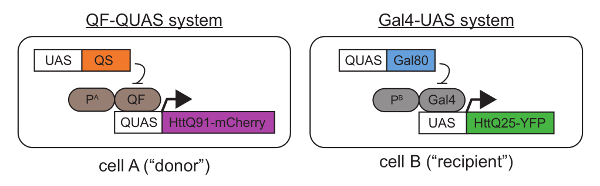
Figure 1. Genetic approach for coupled expression of mutant and wild-type Htt transgenes using the QF-QUAS and Gal4-UAS binary expression systems. In "cell A," an mCherry-tagged mutant Htt protein containing a pathogenic-length polyQ stretch (Q91) is expressed using a QF driver located downstream of a tissue-specific promoter A ("PA"). In "cell B," a YFP-tagged wild-type Htt containing a normal polyQ stretch (Q25) is expressed via a Gal4 driver controlled by tissue-specific promoter B ("PB"). In Figures 2-4, Or67d-QF was used to drive QUAS-HttQ91-mCherry expression in DA1 ORNs, and repo-Gal4 was used to express UAS-HttQ25-YFP in all glia27. Importantly, HttQ91-mCherry is only expressed in QF-expressing cells by virtue of the QUAS sequence placed upstream of the transgene. Similarly, HttQ25-YFP is only expressed via Gal4, which specifically recognizes the UAS. If any overlap in the tissue distribution of the QF and Gal4 drivers is detected, transgenes encoding QS in Gal4-expressing cells and Gal80 in QF-expressing cells can be introduced. Appending fluorescent protein tags onto wild-type and mutant Htt allows for differentiation of the two proteins during imaging and the ability to measure FRET between appropriate donor/acceptor pairs (e.g., CFP/YFP or YFP/mCherry). Please click here to view a larger version of this figure.
2. Micro-dissection and Fixation of Adult Drosophila Brains
Note: This dissection procedure has been modified from a previous publication44, and can be used to prepare brains for imaging direct fluorescence signal from Htt-fluorescent protein fusions. Modifications to the procedure that can be made for immunostaining the brains are discussed in the next section.
- Collect the following materials and place on ice: phosphate-buffered saline containing 0.03% Triton X-100 (PBS/T); microcentrifuge tubes containing 970 µL of 4% paraformaldehyde (PFA) fixative solution prepared by adding 200 µL 20% PFA to 770 µL PBS/T; a clear glass dish containing well; a disposable transfer pipet; two dissecting forceps (one no. 3 and one no. 5).
- Anesthetize the adult flies using CO2 and transfer them to one well of the glass dish on ice.
- Using a transfer pipet, add a small amount (~ 500 µL) of cold PBS/T to the well containing the flies as well as to an empty well. Avoid introducing too many bubbles as they can interfere with the dissection.
- Place the glass dish on a flat surface below a dissecting microscope and adjust the magnification until the fly body fills the field of view and is in focus.
- Position a pair of gooseneck light sources so that light is illuminating both sides of the glass dish. Anchor a folded lab tissue under the glass dish to discard body parts/cuticle during the dissection.
- Using the no. 3 forceps in the non-dominant hand, transfer a single fly into the well containing PBS/T. Immobilize the fly by grabbing hold of its abdomen with the ventral side facing up.
- Keeping the fly fully submerged in PBS/T, remove the fly head with the no. 5 forceps in the dominant hand. Insert one tip of this forceps under the cuticle in the small space adjacent to the proboscis on one side of the fly head. Secure the grip on the head by pinching the forceps to grab hold of the eye from both sides.
- Remove the fly head from its body by pulling the two forceps apart. Dispose of the fly body on a lab tissue positioned nearby. Maintain pressure on the dominant-hand forceps so that the fly head is not lost.
- Position one tip of the no. 3 forceps in the non-dominant hand in the same small space under the cuticle on the other side of the proboscis. Once positioned, pinch the forceps to grip the eyes in the same location on both sides of the head.
- Once the grip on the cuticle is secure, gently pull the forceps apart at 180°. This action will break apart the head cuticle without damaging the brain. Discard cuticle residue on a lab tissue.
- Though ideal, the head cuticle may not be fully removed in one step. In this case, remove the cuticle piece-by-piece until the brain is fully exposed. Take care not to damage the brain by avoiding direct contact with the forceps.
- Remove the dissected brain from the PBS/T either by grabbing ahold of an attached trachea, or by aspirating the brain into the space between the forceps tips by capillary action.
NOTE: Trachea removal is optional, as it tends to not obscure the antennal lobes on the anterior surface of the brain where we typically image. However, if the trachea interfere with imaging, remove them carefully so the brain is not damaged by this manipulation. - Transfer the fly brain into one of the microcentrifuge tubes containing fixative solution on ice. Ensure that the brain detaches from the forceps and is submerged in the fixative solution.
- Once all of the brains have been dissected, subject them to a "short-fix" by placing the closed tubes on a nutator for ~ 5 min at room temperature in the dark.
NOTE: This short fixation step ensures that the brains are easier to handle in subsequent steps and do not adhere to the sides of the microcentrifuge tube. - Remove the majority of the fixative solution with a P1000 pipette and discard it.
- Avoid aspirating brains from the tube during this and any subsequent wash step. Set the P1000 to a lower volume (e.g., 650 µL) and remove the supernatant in two steps. In addition, hold the microcentrifuge tubes in line with a light source (e.g. overhead lights) while aspirating to better visualize the brains.
- Add 1 mL of fresh PBS/T to the brains. Wash quickly (< 1 min) in the dark, aspirate the supernatant from the brains, and discard.
- Repeat the washing with PBS/T in the dark at room temperature according to the following schedule: 2 quick (< 1 min) washes, 1 X 5 min, 3 X 20 min, and 1 X 1 h wash. Secure tops on microcentrifuge tubes and place the tubes on a nutator in between washes.
- If DAPI staining is desired, replace the final 1 h wash with 1 X 30 min incubation with 250 ng/mL DAPI diluted in PBS/T, followed by 2 X quick and 1 X 20 min washes.
- After the last wash, remove the majority of the wash buffer supernatant, taking care not to disturb the brains, and add 30 µL of glycerol-based antifade reagent to the brains. Incubate at 4 °C in the dark without movement for at least 1 h and up to 24 h.
3. Modifications to Section 2 for Immunostaining Adult Brains
Note: Use this protocol for imaging non-fluorescent proteins or for fluorescent protein fusions with weak fluorescence.
- Increase the concentration of Triton X-100 in PBS/T by 10-fold (PBS + 0.3% Triton X-100) for every step. This ensures that enough detergent is present to permeabilize cell membranes and allow antibodies to access intracellular spaces.
- Fix the brains in 4% PFA in PBS/T for 20 min at room temperature.
- After performing all of the washes, incubate the brains in freshly-prepared blocking buffer (PBS/T + 5% normal goat serum (NGS)) for 30 min at room temperature in the dark.
- Remove and discard the blocking buffer. Add 0.5 mL of primary antibody solution per tube prepared as a master mix by diluting primary antibodies as appropriate in fresh blocking buffer (see Table of Materials for commonly used antibodies and dilutions). Incubate the brains in primary antibodies on a nutator for at least 24 h at 4 °C in the dark.
- Remove the primary antibody solution from the brains using a P1000 and reserve in a fresh tube. Add 1 mL of PBS/T to wash the brains.
NOTE: The reserved primary antibody solution can be stored at 4 °C for up to four weeks. We have successfully recycled primary antibodies up to two times in subsequent experiments. - Wash the brains quickly (< 1 min) in PBS/T, aspirate the supernatant from brains, and discard. Repeat this quick wash once more.
- Continue washing with 1 mL PBS/T at room temperature according to the following schedule: 1 X 5 min, 3 X 20 min, and 1 X 1 h wash. Secure tops on microcentrifuge tubes and place the tubes on a nutator during washes.
- After the last wash, remove the majority of the PBS/T supernatant and add 0.5 mL of secondary antibody solution prepared as a master mix by diluting the antibodies as appropriate in fresh blocking buffer (see Table of Materials for commonly used antibodies and dilutions). Incubate brains in secondary antibodies for 24 h at 4 °C in the dark.
- Repeat wash steps in steps 3.5, 3.6, and 3.7 after incubation with secondary antibodies.
- After the final wash, remove the majority of the wash buffer and make sure that all brains are located at the bottom of the tube. Add 30 µL antifade reagent to the brains. Incubate at 4 °C in the dark without movement for 16-24 h.
4. Whole Brain Mounting
- Place a glass microscope slide under a dissecting microscope and label with identifying information.
NOTE: Alternatively, the brains can be mounted directly onto a cover glass to access both sides of the brain during imaging. A protocol describing this mounting procedure has been previously published45. - Remove the brains from each microcentrifuge tube using a blunted pipet tip (prepared using a razor blade to remove the bottom ~ 1 cm off a 1-200 µL tip) and transfer them into the middle of the slide. Transfer as little antifade reagent with the brains as possible.
- Use forceps to position the brains in the desired orientation (e.g., dorsal pointing towards the top of the slide and anterior surface facing up for imaging the antennal lobe). When orienting the brains, consider the light path of the microscope to be used for imaging them.
- Remove excess antifade reagent from the slide using the pointed corner of a folded lab tissue without disturbing the brains. Let the slide sit for ~ 5-10 min in the dark to allow the brains to adhere to the slide.
- Surround the fly brains with four small pieces of broken cover glass placed on each side of the brains to form a square that is ~ 19 mm x 19 mm. Place one edge of a 22 mm x 22 mm No. 1.5 cover glass just outside one of these small pieces of glass, and gently lower the coverslip over the brains to complete the bridge-mount.
- Slowly dispense fresh antifade reagent to fill the surface beneath the coverslip, being careful not to disturb the coverslip or the brains. Wipe off any excess antifade using the pointed corner of a folded lab tissue without directly contacting the coverslip.
- Add a drop of clear, quick-drying nail polish to each of the four corners of the coverslip. Allow to dry for ~ 10 min. Then, seal the four edges of the coverslip with nail polish to completely enclose the brains.
- Image brains immediately, or store at 4 °C until ready.
- If imaging intrinsic fluorescence from Htt-fluorescent protein fusions, image the brains within 24 h for the best signal.
5. Imaging and Quantifying Prion-like Transmission of Aggregates
- Image the mounted brains using a confocal microscope equipped with a 40X or 60/63X oil objective to collect z-slice images through the region of the brain where the selected Gal4 and QF drivers are expressed (Figure 2A, B).
- Excite the fluorescent protein fusions or immunolabeled proteins using the appropriate lasers (e.g., 488 nm for GFP/YFP/FITC or 552 nm for mCherry/Cy3). Set detection windows that capture maximum fluorescent signal while eliminating channel cross-talk using either a spectral detection system or band/long pass emission filters specific for each fluorophore.
NOTE: Be attentive when imaging protein aggregates. It is easy for pixels in the center of each aggregate to become saturated, especially in larger aggregates. Attempt to minimize saturation in the image, but be aware of the risk of losing signal from smaller aggregated species (Figure 2B, C, D). Too much saturation will increase the image background, making it more difficult to identify isolated puncta. Test different imaging settings to optimize before taking the final images in each data set.
- Excite the fluorescent protein fusions or immunolabeled proteins using the appropriate lasers (e.g., 488 nm for GFP/YFP/FITC or 552 nm for mCherry/Cy3). Set detection windows that capture maximum fluorescent signal while eliminating channel cross-talk using either a spectral detection system or band/long pass emission filters specific for each fluorophore.
- Analyze the data by quantifying individual puncta either manually by moving through individual z-slices (e.g., Figure 2C and Figure 4A) or after rendering the confocal slices in 3 dimensions (Figure 3A, B).
NOTE: This can be done in Image J or other image processing software.- If the aggregates are well-separated and there is minimal background fluorescence, use image analysis software to systematically identify, quantify, and analyze the aggregates as distinct "objects" or "surfaces" in a 3D reconstruction of the confocal stack (Figure 3B).
NOTE: Software actions described are specific to the instrument and software used here (see the Table of Materials).- Visualize a confocal z-series in 3D viewing mode. Use the "Analysis" wizard to identify individual spots in a selected channel (e.g. the red channel for HttQ91-mCherry aggregates in Figure 3). Adjust the thresholding and filters to accurately represent all heterogeneously-sized aggregates as individual objects in the image.
- Enable "Split objects" under "Binary Processing Pre-Filter" to separate closely-associated aggregates that are aberrantly merged by the software algorithm. Note that the total number of objects and their associated measurements is reported under "Measurements."
NOTE: This method for quantifying mutant Htt aggregates is not amenable to the immunostaining protocol because the center of amyloid aggregates, is impenetrable to antibodies. As a result, the aggregates appear on the microscope as ring-like structures, and image analysis software is unable to accurately identify and distinguish these individual "spots".
- Quantify wild-type Htt aggregates by manually moving through the z-stack and scoring wild-type Htt aggregates, making sure no aggregates are double-scored if they appear in more than one slice (Figure 4A).
NOTE: This manual quantification approach can be used when the number of aggregates in each confocal stack is reasonable (e.g., 20-50). It is also helpful when image analysis software cannot distinguish individual puncta from surrounding signal in the same channel (e.g., signal from diffuse wild-type Htt in nearby cells) (Figure 2B, C and Figure 4A). Quantifying wild-type Htt aggregates can also be challenging because many normal structures in the brain appear punctate (e.g., cell processes and synapses). Co-localization of wild-type and mutant Htt signals can be used as a selection criterion; however, it is possible that some mutant Htt "seeds" fall below the limit of detection of the confocal. A protein other than Htt that does not aggregate in the recipient cells (e.g., GFP) can be used to label unrelated punctate structures in the brain.
- If the aggregates are well-separated and there is minimal background fluorescence, use image analysis software to systematically identify, quantify, and analyze the aggregates as distinct "objects" or "surfaces" in a 3D reconstruction of the confocal stack (Figure 3B).
- Use image analysis software to perform further characterization of individual aggregates. For example, determine the size distribution of the aggregates (Figure 3C), percent co-localization between mutant and wild-type Htt proteins (Figure 4A), or directly measure protein-protein interactions using FRET (Figure 4B).
- Determine the size distribution of individual puncta using a spot or surface detection algorithm that accurately identifies all visible aggregates in a particular channel. Use the image analysis software to take relevant measurements of the spots or surfaces, for example obtain 'aggregate diameter' (Fig. 3C), 'volume', or 'intensity' information from 'Measurements' in the software "Analysis" wizard described in step 5.2.1.
NOTE: In Figure 3C, we report the distribution of diameters for all HttQ91-mCherry puncta identified in a single DA1 glomerulus. - Determine the percent co-localization between HttQ25-YFP and HttQ91-mCherry aggregates by manually moving slice-by-slice through a confocal z-stack (most accurate method). However, take care not to count any aggregates twice. To prevent this, only count aggregates in a particular z-slice if the plane of focus is through the center of the aggregate (Figure 4A).
- Calculate FRET efficiency for induced HttQ25-YFP aggregates with colocalized HttQ91-mCherry signal using the acceptor photobleaching method.
- First, eliminate potential cross-talk between YFP and mCherry channels by establishing detection windows for mCherry-only and YFP-only signals that produce no signal in the other channel. Using these settings, take a 'before image' of individual HttQ25-YFP puncta and their associated HttQ91-mCherry signal ( Figure 4B).
- Then, photobleach the mCherry signal by adjusting the red laser (e.g., 552 nm) to 100% intensity and scan until the signal is gone. Revert to the settings used for the 'before image,' and take an 'after image' of the same puncta (Figure 4B).
- Calculate fluorescence intensity measurements for each punctum before and after photobleaching using image analysis software.
- Calculate FRET efficiency by subtracting YFP donor fluorescence measured in the 'before image' (YFPinitial) from YFP donor fluorescence in the 'after image' (YFPfinal). Divide this value by YFPfinal, and multiply by 100.
NOTE: FRET efficiency can be calculated pixel-by-pixel or overall for each HttQ25-YFP aggregate (Figure 4B). Acceptor photobleaching is a particularly useful technique for calculating FRET efficiency when the mCherry signal associated with each HttQ25-YFP punctum is sufficiently high to produce detectable YFP dequenching after mCherry photobleaching.
- Determine the size distribution of individual puncta using a spot or surface detection algorithm that accurately identifies all visible aggregates in a particular channel. Use the image analysis software to take relevant measurements of the spots or surfaces, for example obtain 'aggregate diameter' (Fig. 3C), 'volume', or 'intensity' information from 'Measurements' in the software "Analysis" wizard described in step 5.2.1.
The methods described here produce robust data demonstrating prion-like transfer of Htt protein aggregates from one cell population to another in the intact fly CNS. Conversion of wild-type Htt from diffuse to punctate is observed by direct fluorescence of this YFP fusion protein in recipient glia as a result of HttQ91-mCherry expression in donor ORNs (Figure 2A-C and Figure 4A, B). Accurate reporting of prion-like transfer events between these two cell populations requires careful selection of transgenic flies and Gal4/QF drivers to produce strong expression levels of mutant and wild-type Htt transgenes without any overlap during development or in adulthood. In addition, thoughtful design of the fluorescent protein-Htt fusion transgenes can enable powerful downstream analyses. For example, mutant and wild-type Htt aggregates can be quantified as punctate objects either manually (Figure 2C and Figure 4A) or using image analysis software (Figure 3A, B), can be measured and characterized further as aggregate populations (Figure 3C), can be assessed for co-localization between mutant and wild-type proteins (Figure 4A), and can be analyzed for FRET27 (Figure 4B). These analyses require fusion of mutant and wild-type Htt to fluorescent protein tags with sufficiently separated fluorescence properties, but with enough spectral overlap to enable FRET between donor and acceptor pairs (e.g., CFP/YFP9 or YFP/mCherry27).
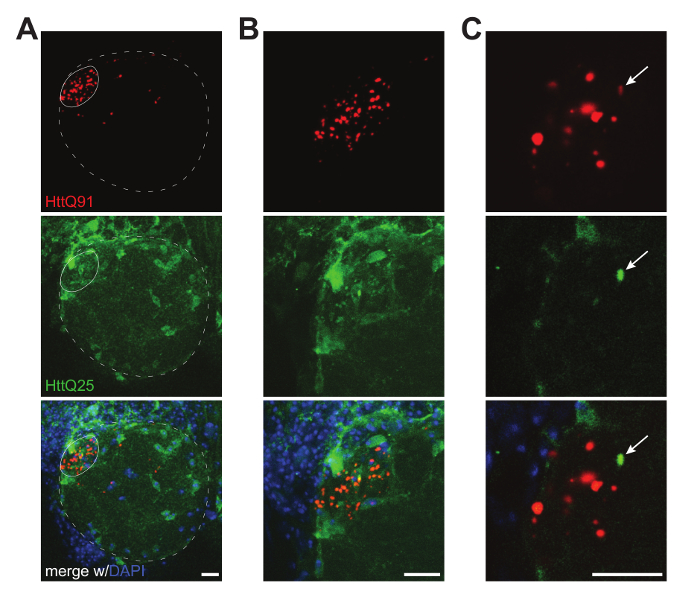
Figure 2. Confocal images of prion-like conversion of glial HttQ25-YFP by neuronal HttQ91-mCherry aggregates. (A) Maximum intensity projection of ~ 30 µm of confocal slices showing one antennal lobe from a male fly expressing HttQ91-mCherry (red) in DA1 ORN axons using Or67d-QF and HttQ25-YFP (green) in glia using repo-Gal4. The approximate boundaries of the antennal lobe and DA1 glomerulus, where DA1 ORN axons terminate, are indicated by the dotted and solid lines, respectively. (B) Maximum intensity projection from ~ 20 µm of confocal slices showing a magnified view of the DA1 glomerular region from A. (C) A single 0.35 µm confocal slice showing a single HttQ25-YFP puncta and its associated HttQ91-mCherry signal (indicated by the arrow in each channel). The signal in the red channel was enhanced to visualize co-localization between HttQ25-YFP and HttQ91-mCherry signals. All images were acquired using a 40X 1.4NA oil objective. Scale bars = 10 µm. Please click here to view a larger version of this figure.
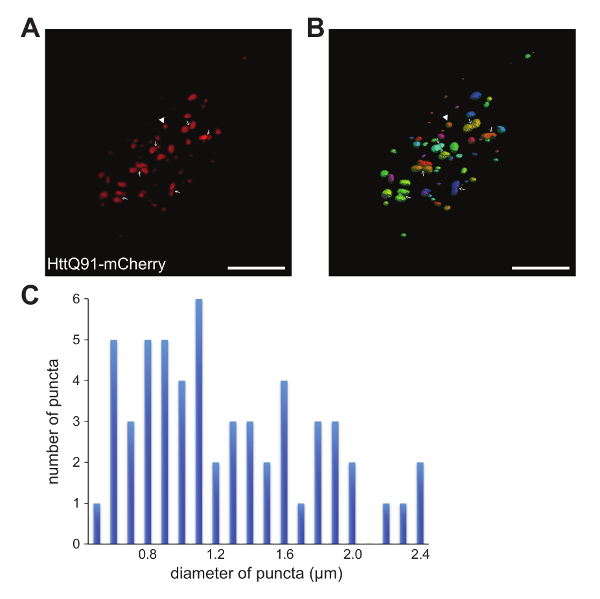
Figure 3. Three-dimensional analysis of HttQ91-mCherry aggregates in DA1 ORN axons. (A) A 3D depiction of HttQ91-mCherry aggregates expressed in the DA1 glomerulus via Or67d-QF using the same data shown in Figure 2B. (B) A screenshot showing individual objects or "spots" identified from the raw data in (A) using an image analysis software package. The software identified 56 objects of varying sizes in this channel/image. The spot indicated by the arrowhead in (B) was measured to have a diameter of ~ 1.2 µm. Arrows point to locations where two objects are inaccurately merged into one spot by the software, likely due to close proximity of the individual puncta. To overcome this, different thresholding settings should be tested in the software and/or merged spots should be separated manually if possible. Scale bars = 10 µm. (C) Histogram showing the distribution of diameters measured by the software for the HttQ91-mCherry "spots" shown in (B). Please click here to view a larger version of this figure.
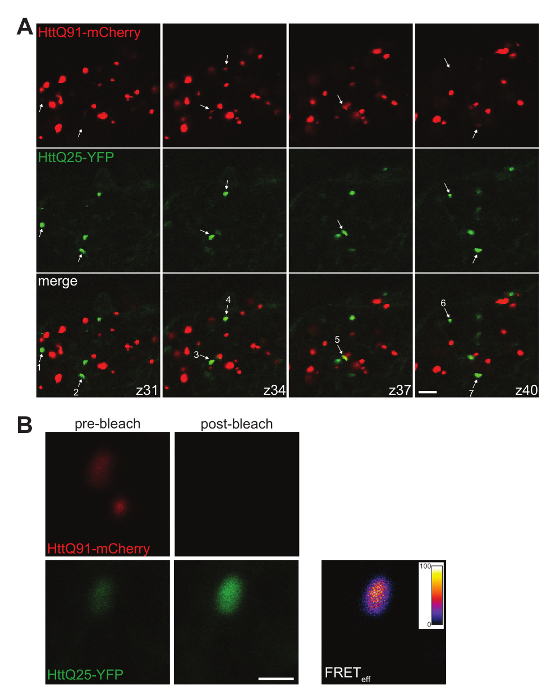
Figure 4. Co-localization and FRET analysis of induced HttQ25-YFP aggregates. (A) A montage of 4 individual 0.35 µm confocal z-slices from a male fly brain expressing HttQ91-mCherry in DA1 ORNs using Or67d-QF and HttQ25-YFP in glia using repo-Gal4. The signals were adjusted so that even small HttQ91-mCherry aggregates are visible and induced HttQ25-YFP aggregates stand out from surrounding diffuse signal. The slices shown are each separated by ~ 1.0 µm (slice number indicated at lower right corner of merged images) so that multiple aggregates can be observed. Arrows indicate HttQ25-YFP puncta that were determined to be in or near focus in that particular z-slice by manually moving through the z-stack. Of the seven HttQ25-YFP puncta indicated here, six have detectably associated HttQ91-mCherry signal (i.e., 86% of the HttQ25-YFP aggregates co-localize with HttQ91-mCherry). Note that the mCherry signal associated with HttQ25-YFP puncta is often weaker than the majority of mCherry-positive puncta in the DA1 glomerulus. Scale bar = 5 µm. (B) A HttQ25-YFP/HttQ91-mCherry-co-localized punctum before (left panels) and after (right panels) mCherry (acceptor) photobleaching. The resulting increase in YFP (donor) fluorescence was used to produce a pixel-by-pixel FRET efficiency (FRETeff) image using the AccPbFRET plug-in for ImageJ46. This particular aggregate has an overall FRETeff of 61%. Scale bar = 1 µm. Please click here to view a larger version of this figure.
