We typically perform liposome purifications on size-exclusion columns. Typical liposome preparations contain a fluorophore of some kind. When liposomes are generated and extruded, the species to be encapsulated are present both inside and outside of the liposomes. By purifying liposomes on a size-exclusion resin (Sepharose 4B), unencapsulated solutes are retained within the pores of the resin, while the larger liposomes are not and elute first (Figure 1A). Collecting fractions and plotting fluorescence vs. fraction number (Figure 1B) typically yields a two-peak trace, with the early-eluting fractions corresponding to the liposomes, which are then collected and used in subsequent applications.
We frequently examine nonenzymatic primer extension reactions, which were a likely means of RNA replication prior to the emergence of ribozyme and protein-based RNA polymerases. These reactions typically employ a fluorescently labeled primer (Figure 2A), which is extended by activated monomers. These reactions can be monitored by gel electrophoresis (Figure 2B) and the resulting electropherograms integrated to obtain rate constants for a given reaction condition (Figure 2C).
To demonstrate that RNA could function inside protocells, we employ hammerhead ribozyme self-cleavage (Figure 3A) as a model RNA catalytic reaction. This reaction requires free Mg2+ to facilitate catalysis, and therefore we used OA/GMO vesicles since they are stable in the presence of 5 mM Mg2+. Similar to primer extension reactions, the hammerhead ribozyme self-cleavage reaction can also be monitored by gel electrophoresis (Figure 3B) and later analyzed to acquire the rate constant under specific conditions (Figure 3C).
We image liposomes employing both fluorescence and transmitted light. Liposomes can be labeled using fluorescent lipids, which give a membrane label (Figure 4A), or using a fluorescent solute within their lumen (Figure 4B). Transmitted light can also be used to observe vesicles (also shown in Figure 4B).
| Table 1.1 pure oleic acid in chloroform | |||
| Component | Stock | Amount | |
| Oleic acid | >99% | 11.7 µL | |
| Chloroform | 1 mL | ||
| Table 1.2 oleic acid and glycerol monooleate (9:1) in chloroform | |||
| Component | Stock | Amount | |
| Oleic acid | >99% | 10.5 µL | |
| Glycerol monooleate | >99% | 1.4 µL | |
| Chloroform | 1 mL | ||
| Table 1.3 oleic acid with 0.2mol% Rhodamine-PE in chloroform | |||
| Component | Stock | Amount | |
| Oleic acid | >99% | 1.6 µL | |
| Rhodamine-PE in chloroform | 10 mM | 20 µL | |
| Chloroform | 1 mL | ||
Table 1. Fatty acid chloroform solutions.
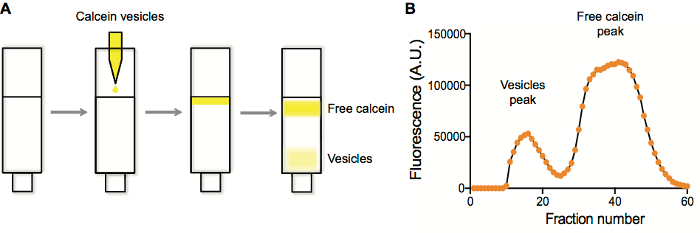
Figure 1. Vesicle purification and fluorescence characterization of purification fraction. A. Separation of vesicles containing calcein from free calcein on a Sepharose 4B column. B. Vesicle and free calcein peak detection by plotting the fluorescence in each well vs. well number after fraction collection. Please click here to view a larger version of this figure.
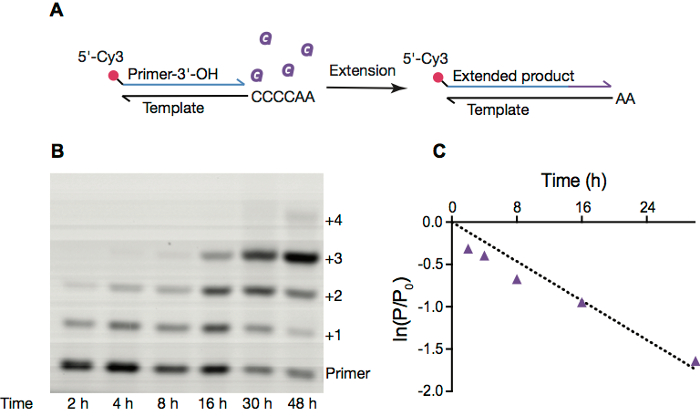
Figure 2. Non-enzymatic RNA replication inside OA vesicles. A. Scheme of non-enzymatic RNA primer extension. B. PAGE image of a primer extension reaction inside pure oleic acid vesicles, with conditions as in section 4. C. Linear fit of the natural logarithm of ratio of amount of primer remaining at given time point to the initial amount of primer vs. time over 30 h. Reaction rate, calculated from the slope of ln(P/P0) vs time, is 0.058 h-1. Please click here to view a larger version of this figure.
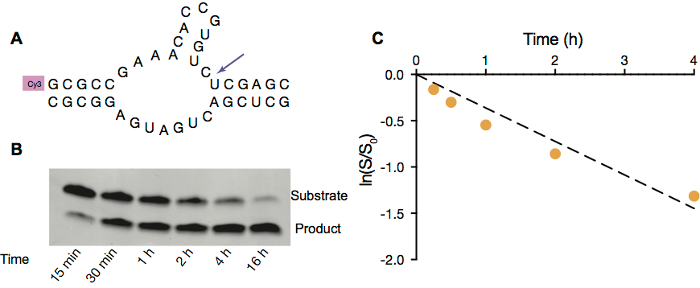
Figure 3. Hammerhead ribozyme cleavage in OA/GMO vesicles. A. Scheme of hammerhead ribozyme cleavage of fluorescently labeled substrate strand (top). B. PAGE image of hammerhead ribozyme cleavage inside OA/GMO vesicles with 5 mM Mg2+. C. Ribozyme activity inside vesicles. Linear fit of natural logarithm of ratio of amount of substrate remaining at given time point to the initial amount of substrate vs. time in first 4 h. Reaction rate, calculated from the slope of ln(S/S0) vs time, is 0.36 h-1. Please click here to view a larger version of this figure.
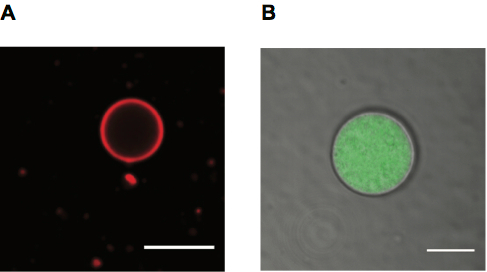
Figure 4. Giant Fatty Acid Vesicles for Microscopy. A. Confocal microscopy image of a Rhodamine PE labeled oleic acid vesicle, scale bar 10 μm. B. Confocal microscopy image of oleic acid vesicle containing Alexa488 labeled RNA with membrane shown in transmitted detector (TD) channel, scale bar 5 μm.
