The Y2H assay has been widely used for finding protein:protein interactions and several adaptations and systems have been developed. For the most part, the same considerations that help ensure success with these previous approaches are important for DEEPN. Some of the important benchmarks include: ensuring expression of DNA-binding domain fusion proteins, ensuring a low background of spurious His+ growth in the diploids containing the bait of interest with an empty prey plasmid, a high mating efficiency of the bait containing MATA yeast with the library-containing MATalpha yeast, and finally, low-stringency conditions that allow the population to grow under conditions that select for many positive Y2H reactions, even ones that produce low levels of His3 activity and a weak Y2H transcriptional response.
One of the critical aspects of DEEPN is to reproducibly introduce the Y2H library into strains that contain different bait plasmids and to reliably select those populations for those library plasmids that create positive Y2H interactions. This becomes more difficult when the complexity of the Y2H library increases since it is harder to ensure adequate transfer of the entire library across different initial populations. In addition, the size of populations chosen to grow under selection conditions and depth of sequencing have to be large enough to observe reproducible changes in the Y2H plasmid composition to reliably identify true positive Y2H interactions. In previous studies, we used commercially available Y2H cDNA libraries. We found the variability in the Y2H library distribution between separate populations carrying the same bait plasmids is low, with an overdispersion between the two initial populations before selection of <0.01 typically, and 0.35 – 0.55 for separate populations after selection4. However, the complexity of some of these commercially available Y2H libraries is fairly low (Figure 7). In addition, many of the clones within it (~60%) are made entirely of cDNA fragments that are 3' of the coding region, which further limits their utility. To demonstrate that the methods above were capable of accommodating more complex Y2H libraries, we created a new Y2H library in the streamlined 'prey' plasmid vector (pGal4AD) containing fragments of genomic DNA from Saccaromyces cerevisiae (strain PLY5725). Genomic DNA was fragmented by shearing, size selected for ranges of 600 – 1500 bp, modified with adaptors, and inserted into pGal4AD to create the SacCer_TAB Y2H library (Figure 8). The library was transformed into PLY5725, which produced a yeast population that was then mated to separate samples of PJ69-4A MATA carrying the pTEF-GBD plasmid alone. Duplicate diploid populations were grown under non-selective and selective conditions. The Y2H plasmid library inserts were analyzed by deep sequencing. When mapped to genomic DNA encompassing 100 bp upstream and downstream of each protein coding region (84% of the whole genome), we found >1.1 million different plasmids in the library for an estimated total size of the library in yeast of ~1.35 million different plasmids. As a comparison, we also transformed a previously described yeast genomic Y2H library11 (PJ_C1,C2,C3 Y2H library) in MATalpha yeast, and subjected it to the same analysis as above. We found that the complexity of our random fragment yeast Y2H library was far higher and had far more plasmids encoding in-frame fragments of each gene than the previously published library (Figure 9). Importantly, the generation of initial library containing diploid populations was very reproducible with an overdispersion of <0.01 (Figure 10). Moreover, the reproducibility of having the two separate yeast populations produce similar redistributions of plasmids after selecting for positive Y2H interactions was very good, yielding an overdispersion of 0.3. Thus, the methods here accommodate larger Y2H libraries of higher complexity than those previously used.
In terms of increasing the ease of DEEPN, we prefer using gene synthesis to make inserts coding for the bait protein of interest. This allows coding regions for protein domains, chimeras, and mutants to be easily incorporated into Y2H bait plasmids, but also allows their codons to be optimized for expression in S. cerevisiae. Expression of two different Gal4-DNA-binding domain fusion proteins is demonstrated in Figure 3. Two different yeast transformants expressing either of two bait fusion proteins were prepared and subjected to SDS-PAGE and immunoblotting with anti-myc antibodies. Note expression levels of bait proteins relative to the Gal4 DNA-binding domain alone from the empty vector. We found it is important not only to verify the expression of the bait fusion protein, but also to verify expression in the exact MATA yeast transformant colony that will be used to expand and mate to the prey library-containing yeast. Once a transformant has been identified, it is possible to freeze it down and store for later use.
Figure 4 shows a test for self-activation where a serial dilution of diploid cells are plated onto CSM-Leu-Trp (+His), CSM-Trp-Leu-His (-His), and CSM-Trp-Leu-His+3AT (-His+3AT) plates and allowed to grow at 30 °C for 3 days. The desired result is to observe growth in the presence but not absence of histidine regardless of whether there is 3AT. This will allow the use of CSM-Trp-Leu-His to select for yeast with a positive yeast 2-hybrid interaction. If there is growth on CSM-Leu-Trp-His plates, then a Y2H selection can still be obtained using the lowest concentration of 3AT that prevents growth. We find that if growth can be blocked in CSM-Trp-Leu-His + 0.1 mM 3AT, then the DEEPN assay can proceed using this condition to select for Y2H interactions. If, however, inhibition of the growth of diploids containing pGal4AD or other empty prey plasmid and the pTEF-GBD-bait fusion plasmid requires higher concentrations of 3AT, it will compromise the performance of the DEEPN procedure and a different bait plasmid should be sought. Note that His+ growth is the only selection for a positive Y2H interaction. We have found this is sufficient to enrich for Y2H interactions in batch.
One of the most critical procedures in the DEEPN workflow is to achieve high efficiency mating of the MATA yeast carrying the bait plasmid with the MATalpha yeast carrying the Y2H prey library. We found that some strains (e.g., Y187)18, housing commercially available cDNA libraries, have relatively poor mating efficiency. For that reason, we engineered a strain to carry Y2H libraries. This strain is based on BY4742, a derivative of S288c. This strain lacks GAL4, GAL80, TRP1, LEU2, and HIS3. It contains no reporters that are responsive to a hybrid Gal4 protein nor a different hybrid (e.g., LexA-VP16). Instead, the source of Y2H-induced His3 production is housed within the MATA strain that would carry the particular bait plasmid. This simplifies the system and allows greater flexibility in that one can use the same library-containing MATalpha cells to mate with a strain expressing a LexA-bait fusion protein and a LexA(UAS)-HIS3 reporter or a Gal4-DBD-bait fusion protein and a Gal4(UAS)-HIS3 reporter.
Once the diploid populations carrying both a bait plasmid and the library are generated, they are diluted and allowed to grow under conditions that just select for both plasmids (e.g., CSM-Trp-Leu) or for both plasmids and a positive Y2H interaction that drives production of His3 (e.g., CSM-Leu-Trp-His). It is important to start with a large amount of the starting population to avoid an evolutionary 'bottleneck' that can skew the population that results after selection for a positive Y2H interaction. Thus, our procedure specifies using 20 mL of out of the 500 mL of diploid population culture to be grown in 750 mL of fresh CSM-Leu-Trp-His media to avoid this problem. With the pTEF-GBD as the bait plasmid, we found that a single round of dilution and growth was sufficient to evolve an informative population. Using different bait plasmids previously, we used two rounds of dilution and growth, an initial 20 mL of into 750 mL, and then 2 mL of from that saturated culture diluted in 75 mL. Figure 6 shows the results from PCR amplification across the library inserts for the diploid population grown under non-selective conditions, as well as a first and second successive round of dilution and growth in selective condition for two different bait plasmids. Note that with no selection, there is a generalized smear of PCR products indicative of a complex and relatively well-normalized mixture of fragments. Upon selection, that pattern changes, with some species greatly enriching so that individual bands can be discerned. Some degree of banding is typical of the successful experiments conducted so far, yet a smear pattern in addition to, or instead of a banding pattern, is indicative of a complex mixture of prey inserts and is desirable if one wants to maximize the number of candidates that DEEPN detects. Very strong banding pattern, where most of the PCR product is found in 1 – 3 bands, indicates that most of sequencing data will be dominated by only 1 – 3 prey inserts. One can compensate for this partly by dedicating more reads from this sample. One can see that if the population is diluted and grown further (see 'selected d2' in Figure 5), the banding pattern is more prominent, indicating that prey plasmids conferring weak but authentic Y2H interactions are being diminished while a select few prey plasmids are increasing their abundance. Successive dilution and growth to this excessive level is deemed counterproductive to the goal of catching the largest number of potential candidates and thus with the protocol here, we recommend one round of dilution and growth be used.
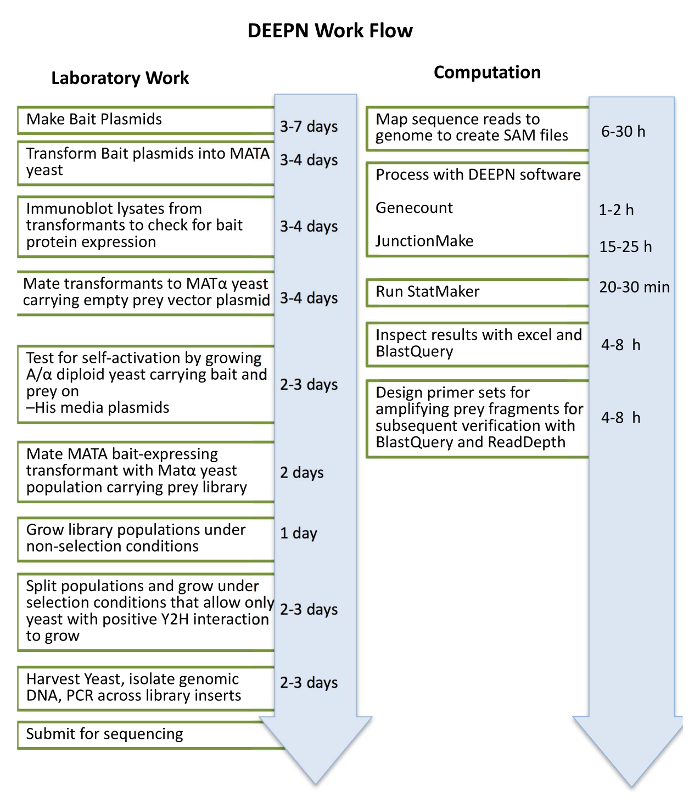
Figure 1: Schematic of DEEPN workflow. The general outline of the laboratory procedures is shown on the left along with the approximate time needed to complete the corresponding task. On the right is the bioinformatics workflow using the DEEPN and Stat_Maker software packages. Please click here to view a larger version of this figure.
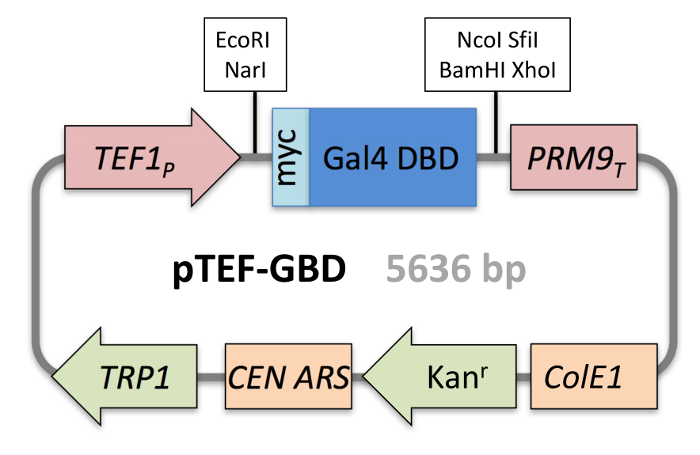
Figure 2: Schematic of pTEF-GBD. The TRP1-containing low copy Gal4 DNA-binding domain fusion protein expression plasmid is shown. It features a constitutive TEF1 promoter, myc epitope tag, the Gal4 DNA binding domain followed a T7 RNA polymerase binding site, polylinker, and PRM9 terminator within a centromere (CEN)-based plasmid. This plasmid also carries Kanamycin resistance and the ColE1 bacterial origin of replication. Please click here to view a larger version of this figure.
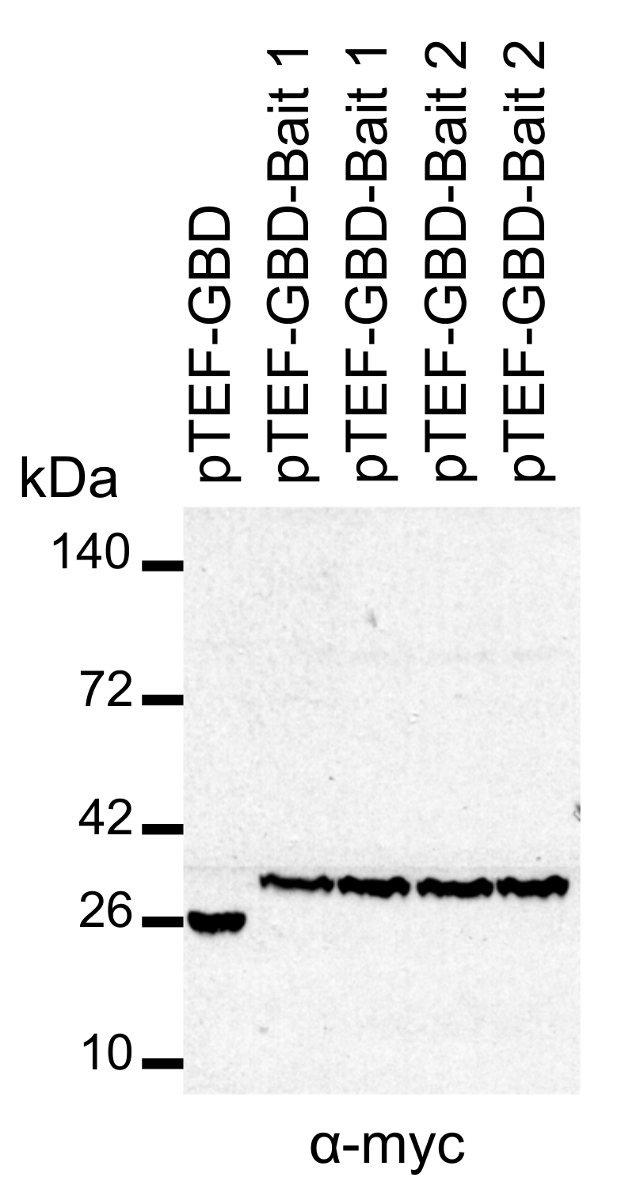
Figure 3: Expression of Gal4-bait fusion proteins. Lysates from yeast expressing different bait proteins fused to the Gal4 DNA-binding domain expressed in pTEF-GBD were subjected to SDS-PAGE and immunoblotting with anti-myc antibodies. pTEF-GBD expresses just the Gal4-DNA-bindng domain alone; pTEF-GBD-bait1 expresses the Gal4-DNA-bindng domain fused to a RhoA lacking its C-terminal prenylation site and housing a mutation locking it into a GTP-bound conformation; pTEF-GBD-bait2 expresses the Gal4-DNA-bindng domain fused to a RhoA lacking its C-terminal prenylation site and housing a mutation locking it into a GDP-bound conformation. Please click here to view a larger version of this figure.
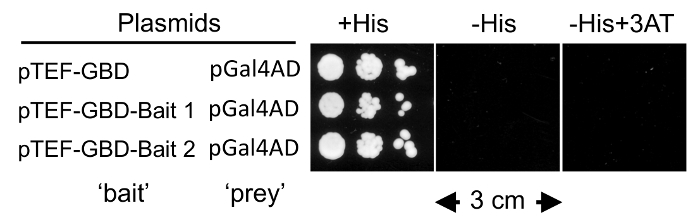
Figure 4: Self-Activation Test. Diploids made from PJ69-4A cells carrying the indicated bait plasmids and PLY5725 cells carrying the indicated prey plasmid were serially diluted and spotted onto CSM-Leu-Trp plates, CSM-Leu-Trp-His plates, and CSM-Leu-Trp-His+3AT plates and grown for 3 days at 30 °C. The PJ69-4A transformants used for mating were the first of each pair shown in Figure 3. Please click here to view a larger version of this figure.
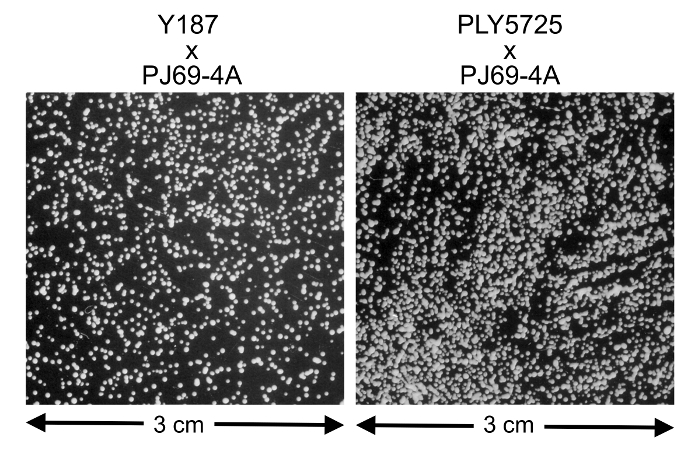
Figure 5: Mating efficiency of Y187 vs. PLY5725. The mating reaction between PJ69-4A containing a TRP1-containing bait vector and Y187 or PLY5725 carrying prey plasmid was performed. 1:10,000 dilutions were plated onto CSM-Trp-Leu to select for diploids. The PLY5725 strain shows a higher mating efficiency than the Y187 strain with ~10 fold more colonies produced. Please click here to view a larger version of this figure.
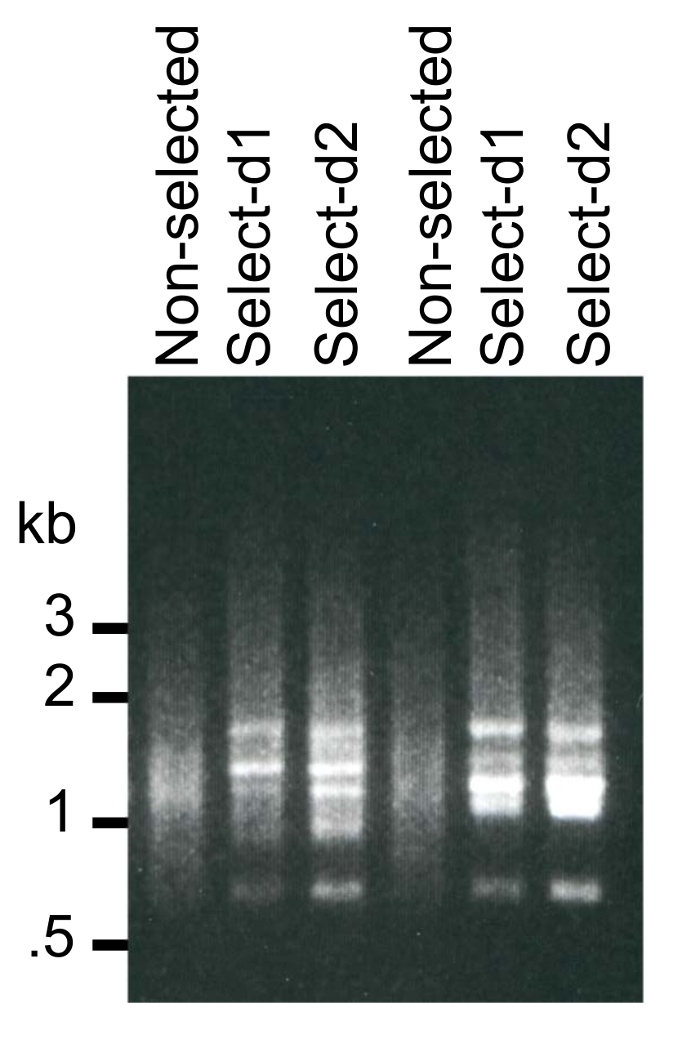
Figure 6: PCR of prey library inserts. DNA was isolated from diploid yeast containing a prey library that was grown under non-selective conditions (CSM-Leu-Trp media) or conditions selecting for a positive Y2H interaction (CSM-Leu-Trp-His media). PCR across the library inserts reveals differences in the repertoire of inserts selected. Two rounds of selective growth were used. An initial round of growth made by diluting 20 mL of the 500 mL of diploid culture into 750 mL of non-selective CSM-Leu-Trp media, and another 20 mL of into 750 mL of CSM-Leu-Trp-His media for an initial round of selective growth (d1). This was followed by an additional round of growth (d2) made by taking 2 mL of the d1 culture and diluting into 75 mL of selective media CSM-Leu-Trp-His. Please click here to view a larger version of this figure.
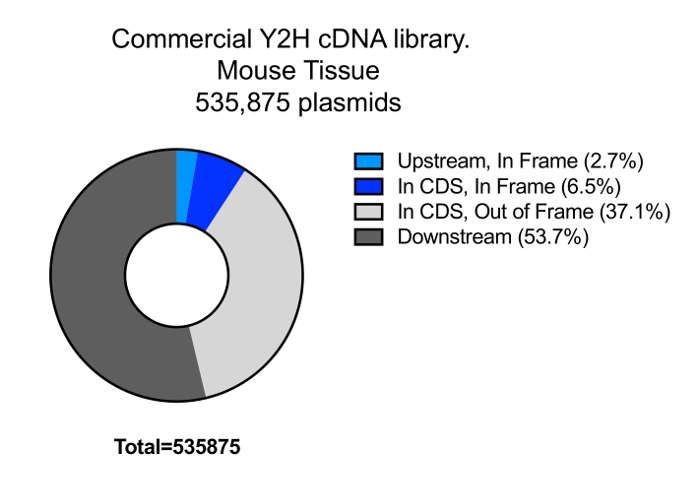
Figure 7: Complexity of Y2H cDNA library. Analysis of the content of a commercially available mouse cDNA Y2H prey libraries: a library from cDNA of mouse brain and one from multiple mouse tissues. Please click here to view a larger version of this figure.
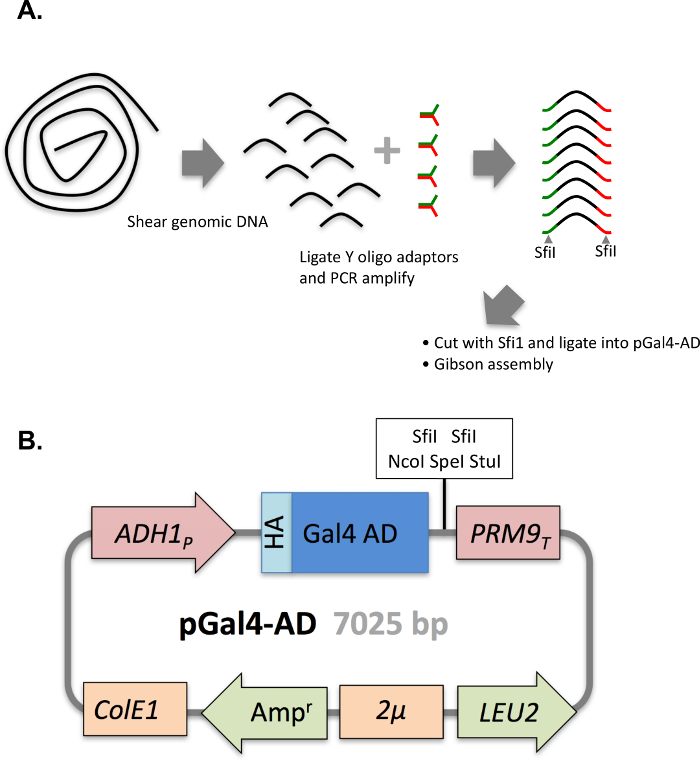
Figure 8: Generation of Y2H yeast genomic fragment library. A. Schematic describing assembly of the yeast genomic fragment library into pGal4AD. Genomic DNA from strain PLY5725 was randomly sheared, ligated with the indicated Y-adaptors, and ligated into SfiI-cut pGal4AD. Ligations were transformed into bacteria to yield 2.2 x 106 independent colonies that were combined and grown prior to isolation of their plasmid DNA to comprise the SacCer_TAB Y2H library. B. The LEU2-containing plasmid (pGal4AD) housing the Gal4 transcriptional activation domain is shown. Please click here to view a larger version of this figure.
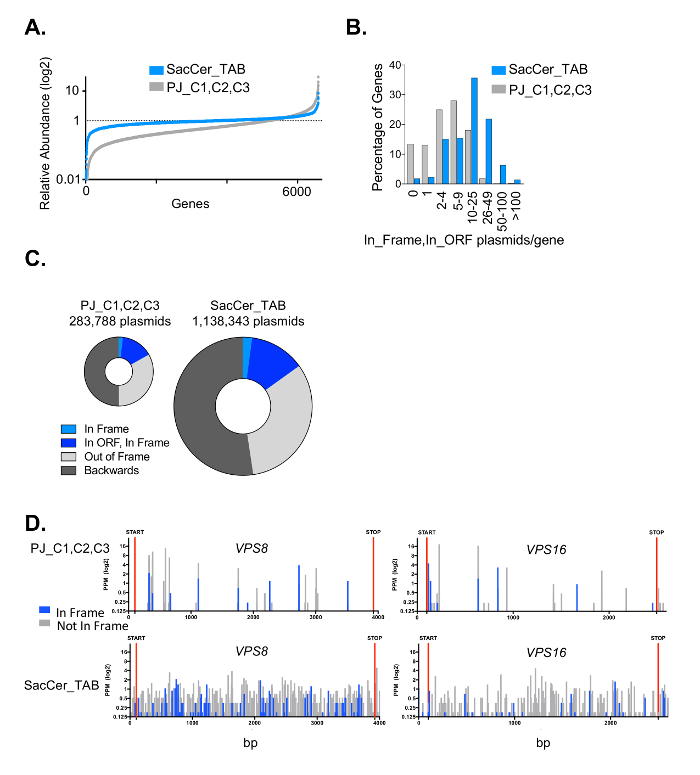
Figure 9: Complexity of Y2H yeast genomic library. The SacCer_TAB genomic library was transformed into the PLY5725 MATalpha strain and the PJ_C1,C2,C3 yeast genomic library was transformed into the Υ187 ΜΑΤalpha strain. Diploids of these populations were made by mating to the PJY69-4A strain. Populations were grown for 10 generations and prey fragments PCR amplified were subjected to high-throughput sequencing and analysis. A. Shows the rank order of reads per each gene in the Y2H libraries divided by the reads per gene found by sequencing the yeast genome. Given equivalent abundance of each gene, each gene would have a value of 1. B. Histogram showing the number of unique plasmids per gene that encode a fragment that is both in the protein coding region (ORF) and in the proper translational reading frame. C. Comparison of the number of different plasmids in each library that encode yeast genes and the proportion of these that are and are not in the correct translational reading frame or are inserted backwards. D. Plots showing positions and abundance of the junctions that map to example genes (VPS8 and VPS16) found for the plasmids in each library. Plasmids with gene fusions that are in the correct translational reading frame are designated blue. Please click here to view a larger version of this figure.
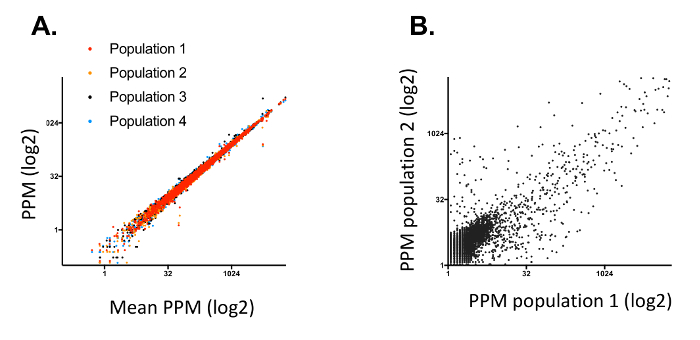
Figure 10: Reproducibility of DEEPN with complex Y2H yeast genomic library. A. Four different yeast diploid populations were created by mating with the SacCer_TAB Y2H library housed in PLY5725, grown under non-selective conditions, and sequenced. The reads per gene for each population individually is plotted as a function of the average rank order value across all populations. B. Shows the reads per gene between two samples after selection for positive Y2H interactions. Please click here to view a larger version of this figure.
