This method, which uses plate-based spectrophotometry to assess GI transit, can be used as a high-throughput replacement of fluorescent microscopy, which is a lower throughput method for assessing the same function (Figure 1). To generate the data in Figure 1, identically treated fish were analyzed for GI transit using either fluorescence microscopy (representative images shown) or spectrophotometry at 4 time points, 0, 4, 8, and 24 h post-dosing; comparison of the data from those experiments gave highly correlated results (linear regression of data r2 = 0.95). The linear regression has a negative slope because microscopy measures the retained fluorescence signal and spectrophotometry measures the transited signal.
The effects of compounds of disparate mechanisms, with well-established GI activity in humans, can be detected in zebrafish using the spectrophotometry assay (Figure 2). Compared to vehicle-treated controls, atropine (4.2 µM) and amitriptyline (5 µM) slowed GI transit, while tegaserod (3.3 µM) and metoclopramide (33 µM) accelerated transit time. Erythromycin (14 µM), expected to accelerate transit time had no effect as measured by this method. Treatment group sizes were 24 before removing data from larvae with no or very low signal. The AUC for the average signal per time point was compared between compound-treated and vehicle-treated groups using Tukey's Honestly Significant Difference for type-1-error control. Effects were considered significant only when p ≤ 0.05. The concentrations used for the above treatments were the maximum tolerated doses, determined in a prior experiment and defined as the highest dose with no observable adverse effect by gross observation.
The spectrophotometry assay can measure dose-dependent effects of compounds. Figure 3 provides data demonstrating that atropine slows GI transit dose-dependently in zebrafish larvae. The lowest dose of atropine tested, 0.042 µM, had no significant effect, while the two higher concentrations each had significant impact, 0.42 µM having less of an effect than 4.2 µM.
A new assay can be assessed by testing positive and negative controls, that is, compounds known to be active and inactive respectively in the target system (in this case the target system is the mammalian GI transit). For the spectrophotometry assay, 18 active (positive) controls and 6 inactive (negative) controls were tested. Based on these experiments, the spectrophotometry assay has high positive predictive value (90.9%), but low sensitivity (55.6%) and negative predictive value (38.5%). These values are derived from the data presented in Table 1. They reflect, in practice, that if the zebrafish transit time is impacted by a treatment, mammalian transit is likely to be impacted. However, if there is no effect on zebrafish transit time, this is not predictive of mammalian effect.
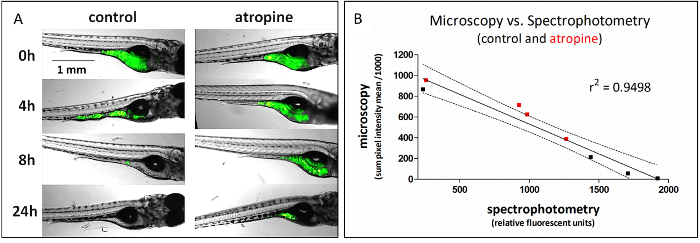
Figure 1: Fluorescent food transit is detected as a loss of signal from microscopic imaging and a corresponding gain in signal by plate-based spectrophotometry. (A) Representative microscopic images from analysis of atropine (4.2 µM) effect on GI transit time. (B) Average signal quantified from microscopic images is highly (negatively) correlated with signal from voided fecal matter (spectrophotometry) from identically treated fish. Data from atropine-treated fish are in red. This figure copied with permission from Cassar et al.11. Please click here to view a larger version of this figure.
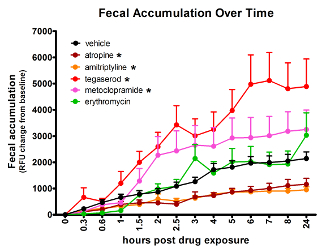
Figure 2: Analyzing fluorescent signal accumulation over time from a multi-well plate allows identification of treatments that change GI transit rate. Asterisk (*) indicates significantly different AUC than vehicle-treated fish. Error bars represent the standard error of the mean signal for larvae in the treatment group per time point. This figure has been reused with permission from Cassar et al.11. Please click here to view a larger version of this figure.
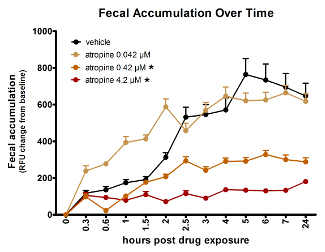
Figure 3: Atropine dose-dependently slows zebrafish gut transit time as reflected by fluorescent spectrophotometry of fecal accumulation over time. Asterisk (*) indicates significantly different AUC than vehicle-treated fish. Error bars represent the standard error of the mean signal for larvae in the treatment group per time point. This figure is reused with permission from Cassar et al.11. Please click here to view a larger version of this figure.
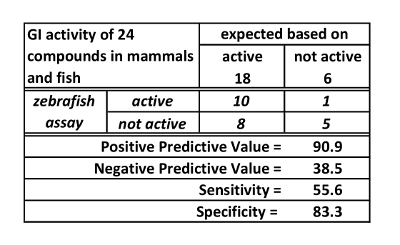
Table 1: GI activity of 24 compounds in mammals and fish. This table is reused with permission from Cassar et al.11.
