1. Preparation of culture medium and culturing of TK6 cells
NOTE: Some chemicals used in this protocol are toxic. Inhaling, swallowing or contacting skin with Cytochalasin B can be fatal. Wear appropriate personal protective equipment including a laboratory coat and two pairs of nitrile gloves. Wash hands thoroughly after handling. Formalin/formaldehyde is toxic if inhaled or swallowed; is irritating to the eyes, respiratory system, and skin; and may cause sensitization by inhalation or skin contact. There is a risk of serious damage to eyes. It is a potential carcinogen.
- Prepare 565 mL of 1x RPMI culture medium. Add 5 mL of MEM non-essential amino acids (100x), 5 mL of sodium pyruvate (100 mM), 5 mL of penicillin-streptomycin-glutamine (100x), and 50 mL of fetal bovine serum (FBS) to a 500 mL bottle of 1x RPMI 1640 medium. Prepare the medium in a biosafety cabinet and store at 2-8 °C. Heat the medium to 37 °C prior to adding it to the TK6 cells (see Table of Materials).
- Thaw 1 mL of TK6 cells (stored at -80 °C in DMSO) in 10 mL of medium. Centrifuge the cells at 200 x g for 8 min and aspirate the supernatant. Transfer the cells to 50 mL of media and incubate at 37 °C, 5% CO2. The doubling time of TK6 cells varies from ~12-18 h and a few (3 or 4) passages will be required for the cells to reach their maximum proliferation rate (see Table of Materials).
- Culture 100 mL of cells to a concentration of ~7-8 x 105 cells/mL.
2. Preparation of clastogens and/or aneugens and Cytochalasin B
- Prepare appropriate stock concentrations of desired clastogens and aneugens. For example, for Mitomycin C, dissolve a full 2 mg bottle in 10 mL of sterile water to achieve a final stock concentration of 200 µg/mL. Mitomycin C can be stored at 4 °C for three months (see Table of Materials).
- On experiment day, prepare dilutions of the desired chemicals that are either 10-fold or 100-fold higher than the desired exposure concentrations if diluting in sterile water or DMSO, respectively.
- For Mitomycin C, prepare 3 mL dilutions in sterile water of 0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 µg/mL. For Colchicine, prepare 3 mL dilutions in sterile water of 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5 µg/mL. Finally, for Mannitol, prepare 3 mL dilutions in sterile water of 5, 10, 20, 30, 40, and 50 mg/mL.
- Prepare a 200 µg/mL stock concentration of Cytochalasin B by dissolving a 5 mg bottle into 25 mL of DMSO. Cytochalasin B can be stored at -20 °C for several months.
3. Exposure of cells to clastogens and/or aneugens
- Add 1 mL of desired chemical (e.g. Mitomycin C) to 9 mL of cells at ~7-8×105 cells/mL in a T25 flask. For the control samples, add 1 mL of sterile water. Place the flasks in a 37 °C, 5% CO2 incubator for 3 h.
NOTE: If chemicals are diluted in DMSO, add only 100 µL of the chemical to each flask and add 100 µL of DMSO to controls. Each flask should contain 9.900 mL of cells. - After 3 h, remove flasks from the incubator and transfer cells to 15 mL polypropylene tubes. Centrifuge at 200 x g for 8 min, aspirate the supernatant and transfer cells to new T25 flasks containing a total of 10 mL of fresh culture medium. Add 150 µL of the stock concentration (200 µg/mL) of Cytochalasin B to each flask to achieve a final concentration of 3 µg/mL.
- Return the flasks to the 37 °C, 5% CO2 incubator for a recovery time equal to 1.5-2.0 doubling times, as recommended by the OECD guidelines9. For the TK6 cells used in this work, the recovery time was 24 h.
NOTE: The doubling time of the TK6 cells used here was 15 h and a recovery time of 24 h (1.6 doubling times) was used. Recovery times less than 1.5 doubling times will reduce proliferation in samples exposed to higher doses impacting the number of BNCs. Conversely, recovery times of more than 2.0 will produce a disproportionate number of polynucleated cells in control samples, skewing cytotoxicity calculations.
4. Preparation of buffers for fixation and labeling of DNA content (see Table of Materials)
- Prepare 75 mM of potassium chloride (KCl) by adding 2.79 g to 500 mL of ultrapure water. Stir the solution for 5 min using a magnetic stirrer and sterile filter through a 200 µm filter. The 75 mM KCl solution can be stored at 4 °C for several months.
- Prepare a sufficient amount of 4% formalin for the experiment, anticipating that a total of 2.1 mL must be added to each sample. For example, to prepare 10 mL of 4% formalin, add 4 mL of 10% formalin stock to 6 mL of 1x Dulbecco's Phosphate-Buffered Saline solution without Ca2+ or Mg2+ (PBS). This 4% formalin can be stored at room temperature for several weeks.
- Prepare 510 mL of wash buffer (2% FBS in 1X PBS) by adding 10 mL of FBS to a 500 mL bottle of 1x PBS.
- Prepare 10 mL of a 100 µg/mL concentration of Hoechst 33342 by adding 100 µL of the stock concentration (1 mg/mL) to 9,900 µL of 1X PBS. The Hoechst 33342 solution can be stored at 4 °C for several months.
5. Sample processing: hypotonic swelling, fixation, cell counting and labeling DNA content
- At the end of the recovery period, remove all flasks from the incubator and transfer all samples to 15 mL polypropylene tubes. Centrifuge all samples at 200 x g for 8 min.
- Aspirate the supernatant, resuspend the cells and add 5 mL of 75 mM KCl. Mix gently by inversion three times and incubate at 4 °C for 7 min.
- Add 2 mL of 4% formalin to each sample, mix gently by inversion three times and incubate at 4 °C for 10 min. This step acts as a "soft fixation".
- Centrifuge all samples at 200 x g for 8 min. Aspirate the supernatant and resuspend in 100 µL of 4% formalin for 20 min. This step acts as a "hard fixation".
- Add 5 mL of wash buffer and centrifuge at 200 x g for 8 min. Aspirate the supernatant and resuspend in 100 µL of wash buffer.
- Transfer all samples to 1.5 mL microcentrifuge tubes.
- Perform a cell count on each sample to determine the number of cells per sample. Samples will be highly concentrated so a 1:100 dilution in 1x PBS (10 µL of sample in 990 µL of PBS) will likely be required to obtain an accurate count.
NOTE: At this point it is best to perform cell counts using a hemocytometer. Adding KCl gives the cytoplasm a translucent appearance, making it difficult for automated cell counters to recognize them. Also, automated counters have difficulty scoring polynucleated cells due to their size. - If not running the samples on the MIFC immediately, they can be stored at 4 °C for several days. When ready to run samples, add 5 µL of 100 µg/mL per 1×106 cells/mL to each sample. Also add 10 µL of 500 µg/mL of RNase per 100 µL of sample for a final concentration of 50 µg/mL. Incubate the samples at 37 °C, 5% CO2 for 30 min.
- Micro-centrifuge all samples at 200 x g for 8 min and use a pipette to remove the supernatant leaving ~30 μL. Use a pipette to resuspend all samples before running on the MIFC ensuring there are no bubbles in the tube. Do not vortex.
6. Starting and calibrating the MIFC
- Ensure the sheath, system calibration reagent, debubbler, cleanser and sterilizer containers are full and the waste tank is empty. Power up the system and double-click on the MIFC software icon. Click the Startup button and make sure that the Start all calibrations and tests checkbox is checked. This will flush the system, load sheath and system calibration reagents, and calibrate the system (see Table of Materials).
7. Running samples on the MIFC
NOTE: This section assumes the use of a 2 camera MIFC. If using a 1 camera MIFC, please see Supplement 1 - Full Protocol, section 7 for the creation of plots during acquisition
- Launch the MIFC data acquisition software (see Table of Materials). Figure 1 shows the instrument settings. Turn on the 405 nm laser and set the laser power to 10 mW (A). Disable all other lasers (including SSC) and set the BF to channels 1 and 9 (B). Confirm that the magnification slider is set to 60x (C), high-sensitivity mode is selected (D), and that only channels 1, 7, and 9 are showing in the image gallery.
- Click on the Scatterplot icon. Select the All population and select Area M01 on the X-axis and Aspect Ratio M01 on the Y-axis. Click on the Square Region icon and draw a region around the single cells. Name this region Single Cells. Right click on the plot and select Regions. Highlight the Single Cells region and change the x-coordinates to 100 and 900 and change the y-coordinates to 0.75 and 1 (Figure 1I).
- Click on the Scatterplot icon. Select Single Cells as the parent population, select Gradient RMS M01 on the X-axis, and Gradient RMS M07 on the Y-axis. Click on the Square Region icon and draw a region around the majority of the cells. Name this region Focused Cells. Right click on the plot and select Regions. Highlight the Focused Cells region and change the x-coordinates to 55 and 75 and change the y-coordinates to 9.5 and 20 (Figure 1J).
- Click on the Histogram icon. Select the Focused Cells population and select Intensity M07 as the feature. Click on the Linear Region icon and draw a region across the main peak in the histogram. Name this region DNA-positive. Right click on the plot and select Regions. Highlight the DNA-positive region and change the coordinates to 2 x 105 and 2 x 106. The range may have to be adjusted depending on intensity peak on the histogram (Figure 1K).
- Set the acquisition parameters (Figure 1E). Specify the file name and the destination folder, change the number of events to 20,000, and select the DNA-positive population.
- Click Load (Figure 1F) and place the control sample into the MIFC. Click the Acquire button to collect the data (Figure 1G). Once the acquisition is complete, click the Return button to return the sample (Figure 1H). Remove the sample tube from the instrument. Repeat this process for all remaining samples in the experiment.
8. Opening a data file in IDEAS
- Launch the MIFC analysis software package (see the Table of Materials). Click on Start Analysis to start the Open File Wizard. Select a data file by browsing to the desired raw image file (.rif). Click the Open button and click Next.
- Since this is a single color assay, compensation is not necessary so click Next to bypass the compensation step. At this stage there is no analysis template to apply, so click Next again. If the analysis template has been downloaded from the supplementary material, select it now. These templates only work with a 2 camera MIFC with BF set to channels 1 and 9 and nuclear imagery in channel 7 during acquisition.
- By default, the .cif and .daf file names are automatically generated to match the .rif. It is not recommended to change the names of the .cif and .daf. Click Next. Set the image display properties by selecting the 01 and 07. Click Next. There is no wizard for this application, so click Finish. It is very important to save the data analysis file (.daf) and the analysis template (.ast) often during sections 9–14 to avoid loss of progress.
9. Creating masks and features to identify BNCs
- Click on the Image Gallery Properties icon (blue/white icon). In the Display Properties tab click Set Range to Pixel Data then change the color to yellow. Click OK. Hoechst images are now easier to view against the black background.
- Create the non-apoptotic cells plot.
- Click on the Analysis tab, then click Masks. Click New then click Function. Under Function choose Threshold, under Mask choose M07 and set the Intensity Percentage to 50. Click OK then OK again. Click Close.
- Click on the Analysis tab, click Features, then click New. For the Feature Type select Area. For Mask select the Threshold(M07,Ch07,50). Click Set Default Name and click OK. Click Close to start calculating the feature values.
- Click on the Dot Plot icon. Select the All population. For the X-axis feature choose the Contrast_M01_Ch01 feature and for the Y-axis feature choose Area_Threshold(M07,Ch07,50). Click OK.
- Click the Square Region button and draw a region around the majority of the cells. Call this region Non-apoptotic. Right click on the plot and click Regions. Highlight the Non-apoptotic region. Set the x-coordinates to 0 and 15 and set the y-coordinates to 50 and 300. Click Close.
- Create the BNC mask (steps 9.3.1-9.3.5) to identify cells that contain only two nuclei.
- Browse for a BNC in the image gallery and click on it. This is to visualize creation of the mask in the Hoechst channel.
- Click on the Analysis tab, then click Masks. Click New then click Function. Under Function choose LevelSet, under Mask choose M07, select the Middle Level Mask radio button, and set the Contour Detail Scale to 3.00. Click OK then OK again.
- Click New then click Function. Under Function, choose Dilate, and under Mask choose LevelSet(M07,Ch07,Middle,3). Set the image to display to Ch07, and set the Number of Pixels to 2. Click OK then OK again.
- Click New then click Function. Under Function choose Watershed, and under Mask choose Dilate(LevelSet(M07,Ch07,Middle,3)2). Set the image to display to Ch07, and set the Line Thickness to 1. Click OK then OK again.
- Click New then click Function. Under Function choose Range, under Mask choose Watershed(Dilate(LevelSet(M07,Ch07,Middle,3)2)). Set the image to display to Ch07. Set the minimum and maximum area values to 115 and 5000, respectively. Set the minimum and maximum aspect ratio values to 0.4 and 1, respectively. Click OK. In the Name field change the text to read BNC then click OK.
- Create the features and plots to obtain the final BNC population
- Spot Count BNC feature: Click on the Analysis tab, then Features, then New. For the Feature Type select Spot Count. For Mask, select the final BNC mask created in 9.3.5. Set the Connectedness to Four and change the name to Spot Count BNC. Click OK then Close to calculate the feature values.
- Spot Count BNC histogram. Click the Histogram icon. Select Non-apoptotic as the parent population. For the X-axis feature choose the Spot Count BNC feature. Click OK. Click on the Linear Region icon. Draw a region across bin 2. Call this region 2N.
NOTE: Refer to section 9 in Supplement 1 - Full Protocol to create the remaining masks, features and plots to identify the final BNC population
10. Creating masks and features to identify MN within the BNC population
- Create the MN mask. Browse for a BNC that contains an MN in the image gallery and click on it. This is to visualize creation of the MN mask in the Hoechst channel. Click on the Analysis tab, then click Masks.
- Create Spot Identification Mask 1:
- Click New then click Function. Under Function choose Spot and ensure the Bright radio button is selected. Under Mask choose M07, set the Spot to Cell Background Ratio to 2.00. Set the Minimum Radius to 2 and the Maximum Radius to 6. Click OK then OK again.
- Click New then click Function. Under Function choose Range, and under Mask choose LevelSet(M07,Ch07,Middle,3). Set the Image to display to Ch07. Set the Minimum and Maximum Area to 80 and 5000, respectively. Set the Minimum and Maximum Aspect Ratio to 0 and 1, respectively. Click OK then OK again.
- Click New then click Function. Under Function choose Dilate, under Mask choose Range(LevelSet(M07,Ch07,Middle,3),80-5000,0-1). Set the Image to display to Ch07. Set the Number of Pixels to 2. Click OK then OK again.
- Click New. Double Click on the Spot(M07, Ch07, Bright, 2, 6, 2) mask to add it to the mask definition. Click the And operator and then the Not operator. Double click the Dilate(Range(LevelSet(M07, Ch07, Middle, 3), 80-5000, 0-1), 2) mask to add it to the mask definition. Click OK.
- Click New, then Function. Under Function choose Range and under mask choose the mask created in 10.1.1.4:
- Select Spot(M07, Ch07, Bright, 2, 6, 2) And Not Dilate(Range(LevelSet(M07, Ch07, Middle, 3), 80-5000, 0-1), 2).
- Set the Image to Display to Ch07. Set the Minimum and Maximum Area to 10 and 80, respectively. Set the Minimum and Maximum Aspect Ratio to 0.4 and 1, respectively. Click OK then OK again. Spot identification mask 1 is complete.
NOTE: Refer to section 10 in Supplement 1 - Full Protocol to create the masks, features and plots to identify the final MN population
- Create Spot Identification Mask 1:
11. Create masks, features and plots to identify the Mononucleated and Polynucleated populations
- Create the POLY mask. Click Analysis, then Masks, then New then Function. Under Function choose Range, under Mask choose Watershed(Dilate(LevelSet(M07, Ch07, Middle, 3), 2)). Set the image to display to Ch07. Set the Minimum and Maximum Area values to 135 and 5000, respectively. Set the Minimum and Maximum Aspect Ratio values to 0.4 and 1, respectively. Click OK. In the Name field, change the text to read POLY then click OK then Close. The Polynucleated cell mask is complete.
- Create the POLY Component Masks.
- POLY Component Mask 1: Click on the Analysis tab, then Masks, then New, then Function. Under Function select Component, and under Mask select the POLY mask. For Ranking Feature select Area, and for Sorting Order click the Descending radio button. Set Rank to 1. Click OK then OK again.
- POLY Component Masks 2, 3 and 4: Repeat all steps in 11.2.1 except set Rank to 2, 3 and 4 to create the individual component masks.
- Spot Count using POLY mask.
- Click the Analysis tab, then Features, then New. For Feature Type, select Spot Count. For Mask choose the POLY mask and set the Connectedness at 4. Click Set Default Name and click OK then Close to calculate the feature values.
- Click the Histogram icon. Select the Non-apoptotic population. For the X-axis feature choose the Spot Count_POLY_4 feature.
- MONO spot count region. Click on the Linear Region icon. Draw a region across bin 1 on the histogram created in 11.3.2. Call this region 1N.
- TRI spot count region. Click on the Linear Region icon. Draw a region across bin 3 on the histogram created in 11.3.2. Call this region 3N.
- QUAD MONO spot count region. Click on the Linear Region icon. Draw a region across bin 4 on the histogram created in 11.3.2. Call this region 4N.
- Identify the MONO population.
- Create the MONO Aspect Ratio Feature. Click the Analysis tab, then Features, then New. Under Feature Type, select the Aspect Ratio feature and under Mask select Component(1, Area, POLY, Descending). Click Set Default Name then click OK.
- Create the MONO Circularity Feature. With the Feature Manager window still open, click New. Under Feature Type, select the Circularity feature and under Mask select Component(1, Area, POLY, Descending). Click Set Default Name then click OK then click Close to calculate the feature values.
- For the circular MONO cells dot plot, click the Dot Plot icon. Select 1N as the parent population. For the X Axis Feature choose Circularity_Component(1, Area, POLY, Descending) and for the Y Axis Feature choose Aspect Ratio_Component(1, Area, POLY, Descending). Click OK. Click the Square Region button and draw a region around the cell population towards the top right portion of the plot. Name this region Circular_1N. Right click on the plot and click Regions. Highlight the Circular_1N region. Change the X Coordinates to 20 and 55 and change the Y Coordinates to 0.85 and 1.0. Click Close.
- Create the Area POLY/Area_M07 feature. Click the Analysis tab, then Features, then New. Under Feature Type, select the Area feature and under Mask select Component(1, Area, POLY, Descending). Click Set Default Name then click OK.
- With the Feature Manager window still open, click New then under Feature Type click the Combined radio button. From the list of features, highlight the Area_Component(1, Area, POLY, Descending) and click the down arrow to add it to the feature definition. Click the division symbol (/). Select the Area_M07 feature and click the down arrow to add it to the feature definition. Click Set Default Name and click OK. Click Close to start calculating the feature values.
- For the final MONO population dot plot, click the dot plot icon. Select Circular_1N as the parent population. For the X Axis Feature choose Aspect Ratio_M07 and for the Y Axis Feature choose Area_Component(1, Area, POLY, Descending) / Area_M07. Click OK. Click the Square Region button and draw a region around the majority of the cells. Name this region Mononucleated. Right click on the plot and click Regions. Highlight the Mononucleated region. Change the X Coordinates to 0.85 and 1.0 and change the Y Coordinates to 0.55 and 1.0. Click Close.
NOTE: Refer to section 11 in Supplement 1 - Full Protocol to create the masks, features and plots to identify the final trinucleated and polynucleated populations.
12. Create a custom view to examine the BNC and MN masks
- Click on the Image Gallery Properties button then click on the View tab. Click on the Composites tab then click New. Under Name type Ch01/Ch07. Click Add Image. Under Image choose Ch01 and set the Percent to 100. Click Add Image again, under Image choose Ch07 and set the Percent to 100.
- Click New and under Name type BNC and MN masks
- Click Add Column. Under Image Type choose Ch01 and under Mask choose None
- Click Add Column. Under Image Type choose Ch07 and under Mask choose None
- Click Add Column. Under Image Type choose Ch07 and under Mask choose BNC
- Click Add Column. Under Image Type choose Ch07 and under Mask choose MN mask
- Click Add Column. Under Image Type click the Composite radio button. The Ch01/Ch07 composite image should be automatically added to the view. Click OK to close the Image Gallery Properties window.
13. Create a custom view to examine the POLY mask
- Refer to section 13 in Supplement 1 - Full Protocol to create a custom view to examine the POLY mask
14. Create a statistics table to enumerate key events
- Click on the Reports tab then click Define Statistics Report. In the new window, click Add Columns.
- Add the BNC count statistic. Under Statistics select Count and under Selected Population choose the BNCs population. Click Add Statistics to add the statistic to the list.
- Repeat step 14.2 to create separate columns for the MN BNCs, MONO, TRI and POLY populations. Click Close then click OK.
- The data analysis template is complete (Figure 2). Save the template (File, Save as Template). The full mask list can be found in Supplement 2 - Mask List.
15. Batch process experiment files using the data analysis template
- Under the Tools menu click Batch Data Files then click Add Batch in the new window.
- In the new window click Add Files to select the experiment files (.rif) to add to the batch. Under the Select a template or data analysis file (.ast, .daf) option, click the open folder icon to browse to and open the data analysis template (.ast file) that was saved in step 14.4.
- Click the Preview Statistics Report button to preview the statistics table. No values will be displayed here since they have not yet been calculated. However, this step serves as a check to ensure the proper analysis template has been selected prior to running the batch.
- Click OK to close the current window. Then click Submit Batches to start the batch processing of all files.
- When the batch processing has completed, a .txt file will be available in the folder that contains all of the .rif files. Use these statistics to calculate genotoxicity and cytotoxicity.
16. Calculating the genotoxicity and cytotoxicity parameters
- Calculating genotoxicity: To calculate genotoxicity, use the statistics table created in 15.5. Divide the number of cells in the MN BNCs population by the number of cells in the BNCs population then multiply by 100:
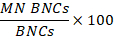
- Calculating cytotoxicity: Determine the total number of POLY cells by summing the number of TRI and QUAD cells.
- Calculate the Cytokinesis-Block Proliferation Index (CBPI) by using the number of cells in the MONO, BNCs and POLY as follows:
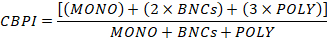
- Finally, calculate the cytotoxicity of each culture by using the CBPI values from the control cultures (C) and chemically exposed culture (T) as follows:
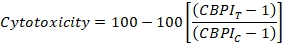
- Calculate the Cytokinesis-Block Proliferation Index (CBPI) by using the number of cells in the MONO, BNCs and POLY as follows:
The analysis method outlined in this paper allows for the automatic identification and scoring of BNCs, with and without MN, to calculate genotoxicity. In addition, MONO and POLY cells are also automatically identified and scored to calculate cytotoxicity. Published scoring criteria6,34 that must be adhered to when scoring these events are implemented in the MIFC data analysis software. The results presented here indicate that statistically significant increases in MN frequency with increasing cytotoxicity can be detected following exposure of human lymphoblastoid TK6 cells to well-known MN inducing chemicals (Mitomycin C and Colchicine). Similar results for additional chemicals tested have been demonstrated in a separate publication32. In addition, results from the use of Mannitol show that non-MN inducing chemicals can also be correctly identified using the MIFC method outlined here. The parameters described in the protocol to create all masks, features and region boundaries will likely have to be adjusted if different cell types (e.g. Chinese Hamster cells) are used to perform the assay.
Figure 3 shows four selected panels to identify BNCs (Figure 3A-3D). Shown here is a histogram that enables selection of cells with two nuclei (Figure 3A) and bivariate plots that enable the selection of BNCs with similar circularity (Figure 3B), similar areas and intensities (Figure 3C) and BNCs that have well-separated, non-overlapping nuclei (Figure 3D) as per the scoring critieria6,34. Figure 3E shows the BF and Hoechst images as well as the BNC and MN masks indicating that BNCs with single or multiple MN can be identified and enumerated. This allows genotoxicity to be calculated by determining the rate of micronucleated BNCs in the final BNC population. Figure 4 shows the application of the Spot Count feature using the POLY mask to identify MONO, TRI and QUAD cells. The number of TRI and QUAD cells can then be summed to obtain the final number of POLY cells (Table 1). This enables cytotoxicity to be calculated by using the formula shown in the protocol. Therefore, each dose point in the experiment can be evaluated by both genotoxicity and cytotoxicity parameters.
Figure 5 shows genotoxicity and cytotoxicity values for the aneugen Colchicine, the clastogen Mitomycin C and for a negative control, Mannitol. For Colchicine (Figure 5A) the 0.02 through 0.05 μg/mL doses produced statistically significant increases in MN frequency, ranging from 1.28% to 2.44% respectively over the solvent control (Table 1). In the case of Mitomycin C (Figure 5B) the two top doses of 0.4 and 0.5 μg/mL produced statistically significant MN frequencies when compared to solvent controls. These MN frequencies were 0.93% at 0.4 μg/mL and 1.02% at 0.5 μg/mL (Table 2). Finally, for Mannitol (Figure 5C), no doses tested induce a cytotoxicity over 30%, nor did they produce significant increases in MN frequency when compared to solvent controls, as expected (Table 3).
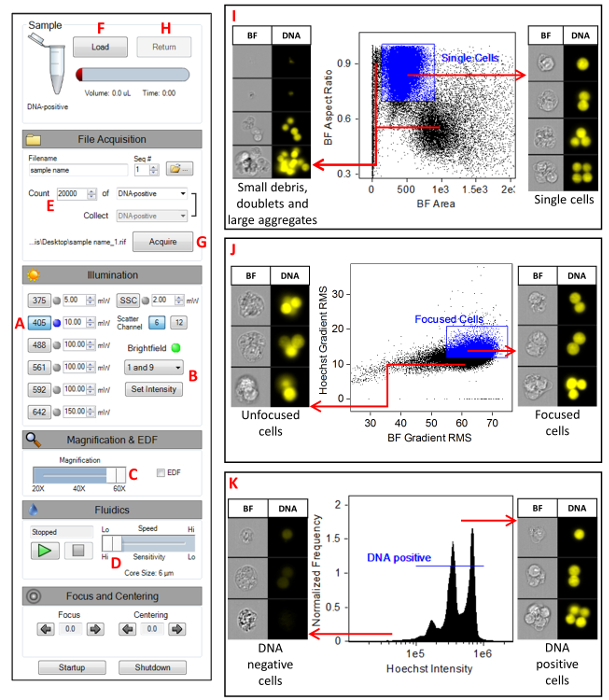
Figure 1: MIFC instrument settings. A screenshot of the MIFC settings as described in section step 7 of the protocol. (A) Setting the 405 nm laser power to 10 mW. (B) Setting the BF channels 1 and 9. (C) Selecting the 60x magnification objective lens. (D) Selecting the slowest flow speed which generates imagery with the highest resolution. (E) Specifying the number of events to be collected to 20,000. (F) Clicking the Load button to begin the sample load process. (G) Clicking the Acquire button to begin acquiring imagery. (H) Clicking the Return button to return any unused sample. (I) Scatterplot of BF Aspect Ratio versus BF Area for the selection of single cells. (J) Scatterplot of Hoechst Gradient RMS versus BF Gradient RMS for the selection of focused cells. (K) Histogram of Hoechst intensity for the selection of DNA positive cells. Please click here to view a larger version of this figure.

Figure 2: Analysis software gating strategy. A screenshot of the gating strategy described in section 9 of the protocol. Regions are shown in sequential order for the identification of binucleated cells (red box), micronuclei (yellow box), and mono- and polynucleated cells (blue box). Please click here to view a larger version of this figure.
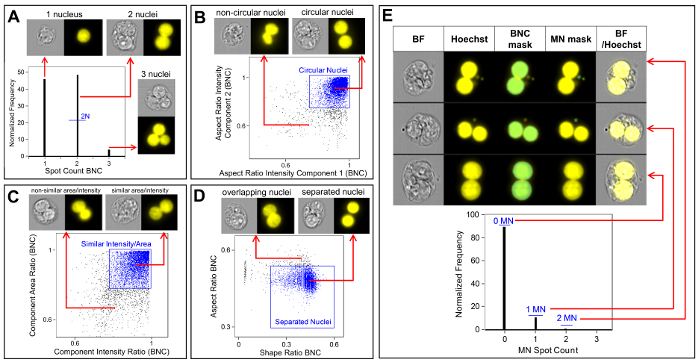
Figure 3: Identification and scoring of BNCs with and without MN. (A) Selection of cells that have two distinct nuclei. (B) Identification of binucleated cells (BNCs) that have two highly circular nuclei through the use of the Aspect Ratio Intensity feature. (C) Selection of BNCs that have nuclei with similar areas and intensities. This is accomplished by calculating the ratio of the area of both nuclei and the ratio of the aspect ratio of both nuclei. (D) Use of the Shape Ratio and Aspect Ratio features to identify BNCs that have two well-separated nuclei. (E) The Spot Count feature using the micronucleus (MN) mask demonstrating that BNCs with single or multiple MN can be identified and enumerated. Please click here to view a larger version of this figure.
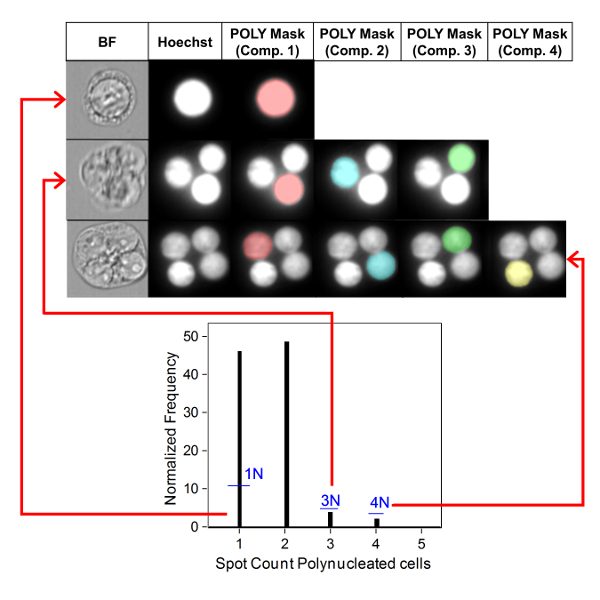
Figure 4: Identification and scoring of MONO and POLY cells. Use of the spot count feature to identify and enumerate mono-, tri- and quadranucleated cells. Component mask 1 allows the identification of mononucleated cells (top image). Component masks 1 through 3 allows the identification of trinucleated cells (middle image). Component masks 1 through 4 allows the identification of quadranucleated cells (bottom image). This figure has been modified from Rodrigues 201832. Please click here to view a larger version of this figure.
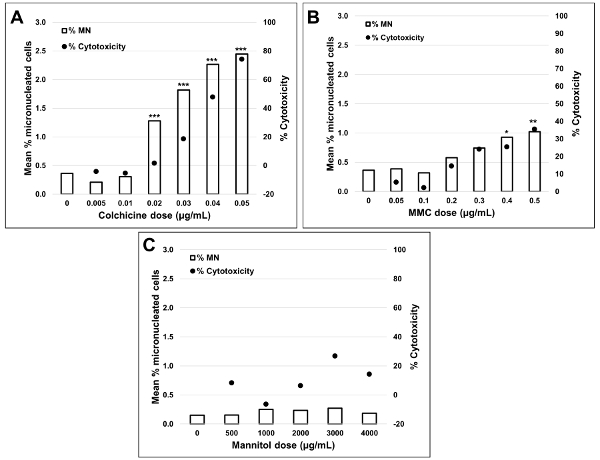
Figure 5: Quantification of cytotoxicity. Cytotoxicity quantified using the cytokinesis block proliferation index (black circles) and genotoxicity quantified using the percentage of MN (clear bars) following a 3 h exposure and 24 h recovery for (A) Colchicine, (B) Mitomycin C and (C) Mannitol. Statistically significant increases in MN frequency compared to controls are indicated by stars (chi-squared test; *p < 0.05, **p < 0.01, ***p < 0.001). All quantities are the average of two replicates at each dose point. This figure has been modified from Rodrigues 201832. Please click here to view a larger version of this figure.
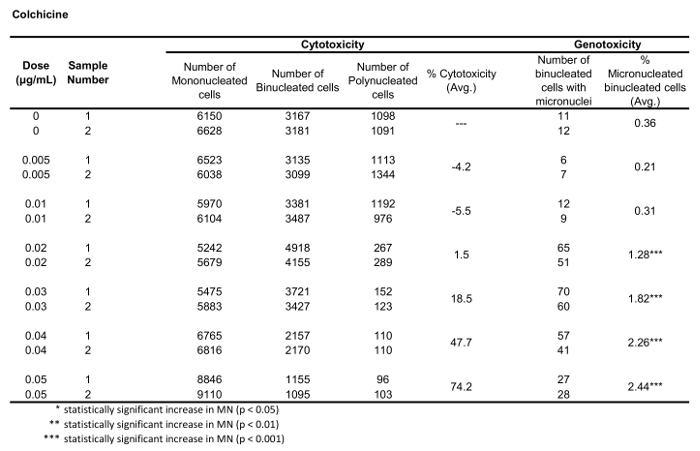
Table 1: The parameters required to calculate cytotoxicity (the number of mono-, bi- and polynucleated cells) and genotoxicity (the number and percentage of micronucleated binucleated cells) for Colchicine. All calculated quantities are the average of two replicates at each dose point.
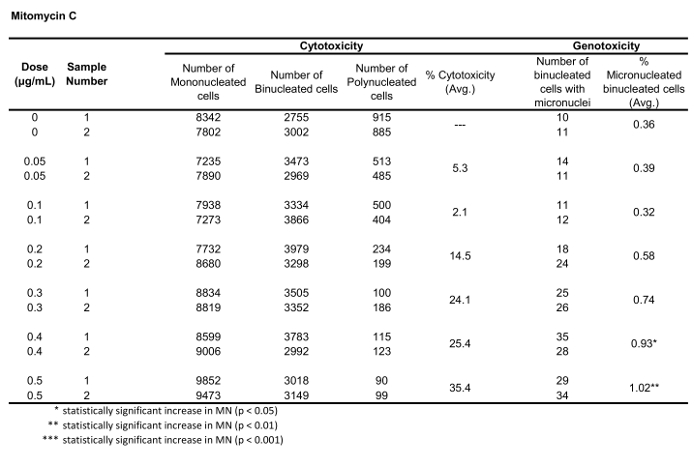
Table 2: The parameters required to calculate cytotoxicity (the number of mono-, bi- and polynucleated cells) and genotoxicity (the number and percentage of micronucleated binucleated cells) for Mitomycin C. All calculated quantities are the average of two replicates at each dose point.
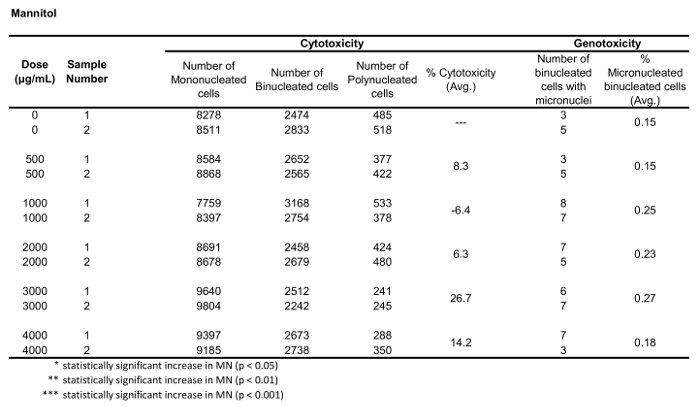
Table 3: The parameters required to calculate cytotoxicity (the number of mono-, bi- and polynucleated cells) and genotoxicity (the number and percentage of micronucleated binucleated cells) for Mannitol. All calculated quantities are the average of two replicates at each dose point.
Supplement 1: Full Protocol. Please click here to download this file.
Supplement 2: Mask List. Please click here to download this file.
