Using the live imaging protocol described above, we capture high resolution image stacks for the subsequent analyses and quantification. Supplementary Video 1 shows the maximum intensity projected (MIP) image series collected from a representative individually labeled LNv. Figure 2B shows the corresponding montage of eight frames of the image series. In both panels, arrowheads mark retraction events and arrows mark extension events.
Next, we perform semi-automated 4D tracking of the branch terminals using an image annotation software (Table of Materials). Figure 3B shows annotations of the dynamic branch terminals from a representative individually labeled LNv. Using this software, we visualize the image stacks in 3D, manually mark all branch terminals and use the Spots module to track the terminals' movements through time. This software generates time-stamped trajectories for every annotated branch terminal, serving as visual representations of the dynamic events on the dendritic arbor.
We then use the coordinate information from annotated branch tips to calculate the direction and distance of branch movements at each time point. Screenshots in Figure 4A,B show how this is achieved. The processed spreadsheets generate output files that we use to quantify dendrite dynamics with four parameters, including percentage of dynamic branches, number of new branches, cumulative distance traveled, and number of branch movement events. Figure 4C shows comparisons of dendritic branch dynamics for individually labeled LNvs from different developmental stages. Consistent with the findings in mammalian neurons and zebrafish tectal cells, the LNv dendrite dynamics are developmentally regulated14,15. LNv dendrites in younger larvae, 48-72 h after egg laying (AEL), are significantly more dynamic compared to those in older larvae, 96-120 h AEL, as measured by all four parameters, and the developmental transition from the dynamic to stable state occur between 72 to 96 h AEL8.
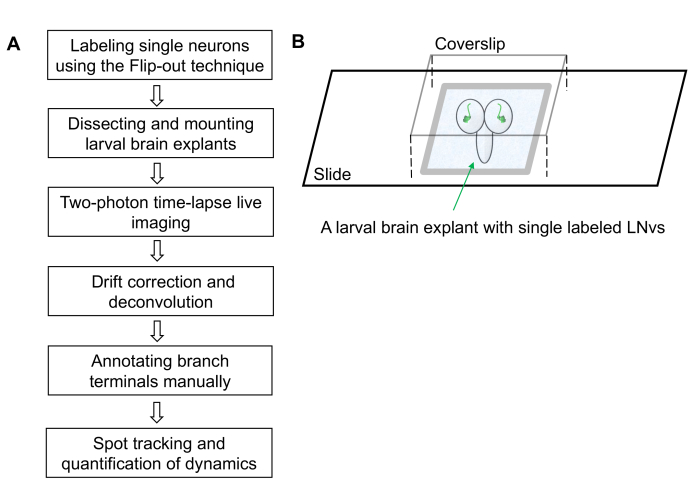
Figure 1: Workflow of the dendrite dynamics imaging and quantification protocol. (A) The protocol contains six steps covering sample preparation, image collection, image processing and semi-automated quantification of dendrite dynamics. (B) A schematic diagram illustrating an imaging chamber containing a larval brain explant mounted with the dorsal side up. The brain explant has an individually labeled LNv in each lobe and is immersed in the external saline solution. The vacuum grease barriers support the weight of the coverslip and forms a small chamber that prevents the brain from moving. Please click here to view a larger version of this figure.
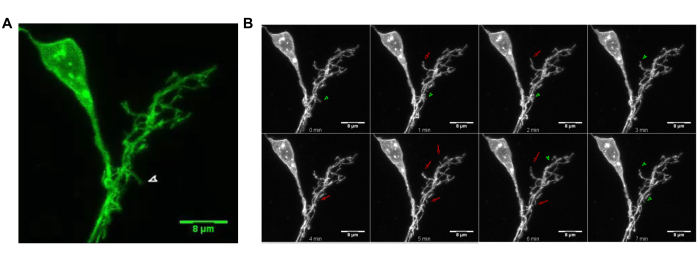
Figure 2: Time-lapse imaging of dendritic branch dynamics in 3D. (A) The dendritic arbor of an individually labeled LNv from a 3rd instar larva. (B) The corresponding montage image of (A) illustrating the representative branch movements. Arrowheads (Green) mark retraction events; arrows (Red) mark extension events. Please click here to view a larger version of this figure.
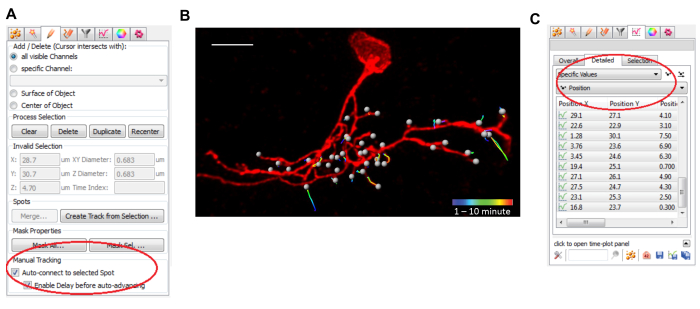
Figure 3: Manual annotation and automatic tracking of dendritic branch terminals in an image annotation software. (A) A screenshot shows the settings for branch terminal tracking using the Spots module. The red oval outlines the Manual Tracking option. (B) Representative time-stamped trajectories of annotated branch terminals (yellow spots) generated by the image annotation software in an individually labeled LNv. Scale bar = 5 µm. (C) A screenshot shows the interface for viewing and exporting the coordinate information from the Spots module. Figure 3B has been modified from Sheng, C. et al. 20188. Please click here to view a larger version of this figure.
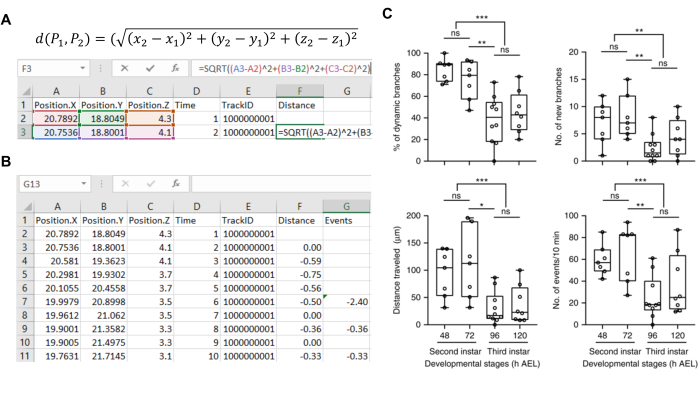
Figure 4: Quantification of dendritic branch dynamics using the coordinate information of the branch terminals. (A) A screenshot shows the spreadsheet containing the X, Y and Z coordinates, Track IDs and formula used for calculating displacement of spots in the adjacent frames. (B) A screenshot shows representative results obtained from tracking one branch. The minor movements (<0.3 µm) were filtered out. The retractions are marked by assigning their value to negative (Distance column). By summing up adjacent positive/negative values, the displacement of each biological extension/retraction is computed (Events column). (C) Representative quantification of results showing the developmental regulation of dendrite dynamics. Data are presented as a box plot (box, 25-75%; center line, median) overlaid with a dot plot (individual data points). Figure 4C has been modified from Sheng, C. et al. 20188. Please click here to view a larger version of this figure.
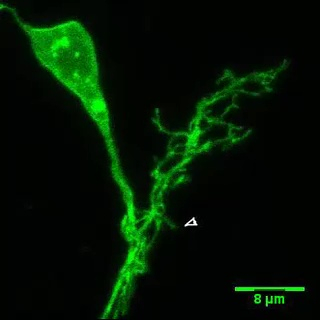
Supplementary Video 1: Ten maximum-intensity-projection (MIP) images played at a speed of 2 frames per second. The whole dendritic arbor was imaged at 1 min per Z-stack. Please click here to view this video (Right click to download).
Supplementary File 1: Batch Column Filtering.R Script. Please click here to view this file (Right click to download).
Supplementary File 2: Batch Column Sum.R Script. Please click here to view this file (Right click to download).
