Here, we present an approach to study plasmid evolution by quantifying plasmid persistence in a population. First, we show how to build the plasmid carrying E. coli strain MG1655 pCON that is subsequently introduced to an evolution experiment. Second, we present a straightforward method to follow plasmid abundance in the evolving bacterial populations. Finally, we show how to visualize plasmid molecule size and conformation.
In our previous work8 using the presented approach we conducted an evolution experiment following the persistence of antibiotic resistance plasmids in E. coli in the absence of antibiotics (Figure 4). Our representative results show the evolution of populations at 37 °C and a dilution rate of 10-4. Following the abundance of plasmid-carrying cells, we observed a decrease in the frequency plasmid-carrying host cells over time (Figure 4). Our approach enabled us to discover that the plasmid loss was a result of the condensed plasmid genome architecture, which led to plasmid instability caused by conflicts introduced by transcription of the resistance gene and replication of the plasmid itself. Visualizing the plasmid molecules enabled us to discover that these conflicts led to an unstable plasmid conformation (i.e., plasmid multimer formation, Figure 5). Nonetheless, we observed plasmid stability evolution without the exposure to antibiotics (Figure 4). The evolved stability was conferred by a plasmid intrinsic duplication that abolished the transcription-replication conflicts and led to the formation of stably inherited plasmids. Our results thus demonstrate the importance of recombination and genome amplification in adaptive evolution of genetic elements.
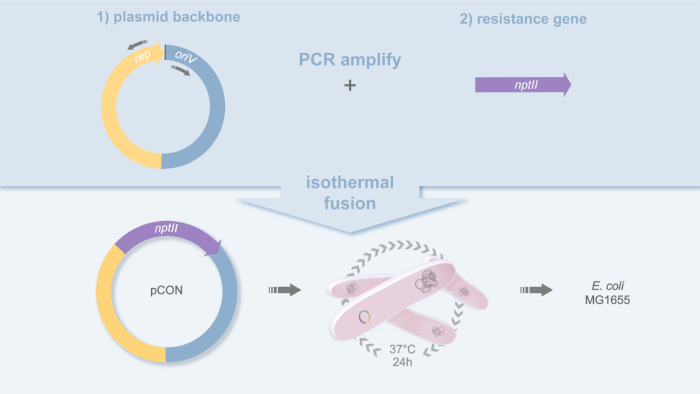
Figure 1: Plasmid design of pCON. Schematic representation of the cloning strategy used to build the plasmid pCON. The plasmid backbone (pBBR1) and antibiotic resistance gene (nptII) are PCR amplified and fused by isothermal fusion20. This yields the plasmid pCON and the strain MG1655 pCON. Please click here to view a larger version of this figure.
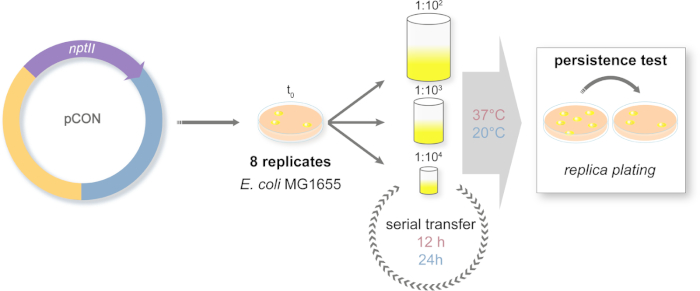
Figure 2: Design of the long-term evolution experiment. Schematic representation of the serial transfer experiment. The plasmid-carrying (pCON) populations are plated on selective media. Ancestral colonies are randomly chosen from the plate and introduced to a serial transfer system. The transfers are conducted with three different dilution approaches to simulate population bottlenecks of different sizes. The dilutions are serially repeated. The experiment is conducted in two temperature regimes. The plasmid-host frequency is measured along the experiment via replica plating. Please click here to view a larger version of this figure.

Figure 3: Replica plating. Schematic representation of the steps used in replica plating of bacterial populations. Please click here to view a larger version of this figure.
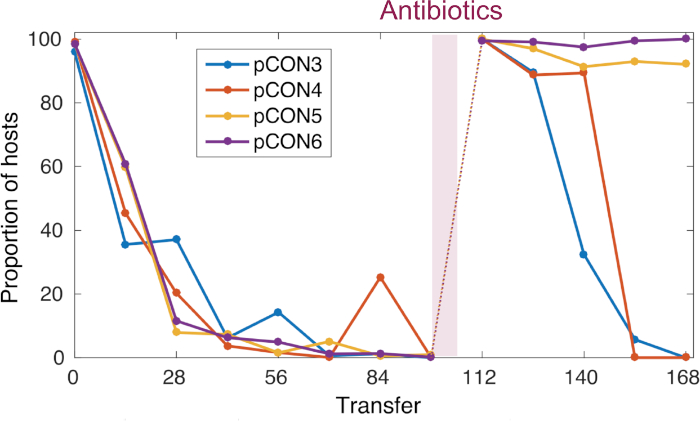
Figure 4: Representative pCON frequency over time. pCON persistence is shown as the proportion of hosts (representative replicate populations) during the evolution experiment. For 98 transfers, pCON plasmid populations evolved under nonselective conditions with a dilution factor of 10-4. All replicates carrying the plasmid pCON decreased in the population. Afterwards, the populations were exposed to antibiotics for overnight incubation and were cultivated again under nonselective conditions to test for plasmid stability evolution. This figure has been modified from Wein et al.8 Please click here to view a larger version of this figure.
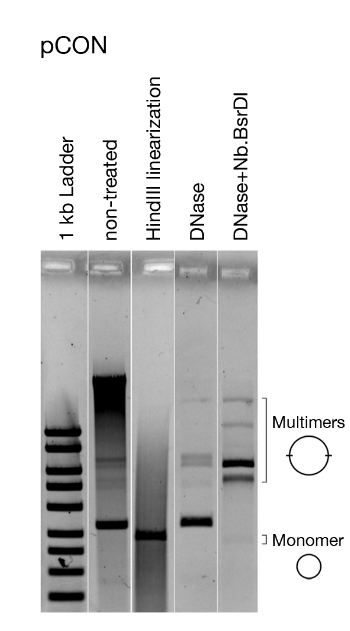
Figure 5: Representative analysis of the plasmid conformation. Visualization of the model plasmid pCON. Visualized is the untreated plasmid DNA directly after extracting, linearized plasmid DNA, and treated with DNase that only cuts chromosomal DNA as well as enzymatically nicked DNA (i.e., open circle plasmid DNA). Linearizing the plasmid shows that all plasmids are of the same size. Removing chromosomal DNA and nicking pCON reveals the presence of dimers and other multimers. This figure has been modified from Wein et al.8. Please click here to view a larger version of this figure.
