All experiments with mice were approved by the UAB Institutional Animal Care and Use Committee (IACUC).
1. Plate Preparation
NOTE: Coverslips can be used to image DP cells at the end of the assay. Be sure the plate lid is on during all incubation and rinsing steps outside of the sterile tissue culture hood to prevent contamination during sample processing.
- Coverslip preparation
- Autoclave circular coverslips.
- Filter ultrapure water under the hood.
- Transfer the coverslips to a 24-well plate and cover each one with 400 μL of 0.1 mg/mL poly-D-lysine.
- Allow the coverslips to soak, immersed for 5 min, on a rocker at 12 rpm or orbital shaker at 40-50 rpm. Be sure lid is on to prevent contamination.
- Rinse the coverslips with filtered ultrapure water for about 5 min on a rocker at 12 rpm or shaker at 50 rpm. Repeat.
- Allow the coverslips to dry for at least 2 h under the hood with the plate lid off to facilitate evaporation. These coverslips should be used within 48 h.
- Seed DP cells atop coverslips and process at the end of the assay for immunofluorescence (section 3.3).
- Coating filters
- Dilute laminin to 10 μg/mL. Filter the diluted laminin under the hood.
- Pipette 450-500 μL of 10 μg/mL laminin into each well of a 24-well plate.
- Place transwell filter, 3 μm porosity, in a well so that it contacts the laminin solution and leave it in a 37 °C incubator for either 2 h or overnight. Filter pores should allow some of the solution to diffuse and coat both the top and bottom of the filter. These filters should be used within 48 h or be refrigerated at 4 °C for up to 1 week.
2. Cell Plating with Optional Genetic Manipulation
- Mice: Genetic alteration of DP cells can be performed (but is not required) to study mesenchymal-neuronal interactions. See sections 2.3 and 2.4 for suggested genetic variations.
- DP dissection, dispersion and plating (Figure 1)
NOTE: For DP dissection, use ultra-fine, straight-edge forceps. The ultra-fine edges will allow the user to wedge the forcep edge between the mineralized structure and DP tissue.- Harvest P5-P8 mice. At this stage, the teeth should be mineralizing, and the root is open.
- Anesthetize the neonates via hypothermia by placing them in a dish in the 4 °C refrigerator until they no longer move or respond to touch. Euthanize neonates by decapitation and in accordance with IACUC procedures at the designated facility.
- Prepare a 3-5 mL aliquot of 0.25% trypsin-EDTA in a 50 mL conical tube to collect the DP from each mouse. This will facilitate the digestion of dental pulp from postnatal mice. Use more than 3 mL if digesting tissue from more than 10 postnatal mice.
- Place the head on a disposable underpad so that the mouth is toward the ceiling and the base of the neck is flat on the work surface. Use a razor blade in a sawing motion to separate the mandible from the maxilla.
- Optionally remove the tongue either with scissors or with forceps to allow easier access to the molars.
- Place the opened head in a dish atop a sterile gauze pad and place the specimen under a dissecting microscope (Figure 1D).
- Remove the alveolar bone tissue surrounding first molars. Submandibular teeth are not fully erupted at this point. Insert forceps into alveolar opening and tease the tissue away from the tooth toward the buccal (cheek) or lingual (tongue) side of the mouth. Maxillary teeth will require full removal of the cleft around the tooth for exposure and removal.
- Gently transfer the submandibular and maxillary first molars (M1s) to a separate cell culture dish with 1x phosphate-buffered saline (PBS).
- Repeat 2.2.4-2.2.8 until all M1s are collected. Keep the dish containing the M1s on ice during the harvesting.
- Remove the Enamel Outer Organ (EOE) surrounding the outside of each M1. This can alternatively be done after step 2.2.11.
- With a set of forceps, rotate the M1 so the cusps are down and the open root is exposed. There will be an oval opening on the bottom of the tooth, and opaque DP tissue encapsulated by a thin layer of dentin and enamel.
- Using the tip of the forceps, gently loosen the DP by running one arm of the forceps around the internal circumference of the mineralized tissue. Remove DP tissue out of the mineralized structure and transfer it to a third dish containing 1x PBS. Remove the EOE if it was not already separated (Figure 1E).
- Transfer all DP tissue to 0.25% trypsin-EDTA in a 50 mL conical tube. Vortex the mixture and place in a 37 °C warm water bath for 10 min. This can be done with the same forceps or with long, vial forceps. The tissue will be difficult to disperse and will require vortexing every 3-4 min. Do NOT exceed 10 min trypsinization since the trypsin can damage cell membranes.
- Under a sterile hood, add warmed co-culture media (Table 1) to a final ratio of at least 1:1 media to trypsin to inactivate the enzyme. Larger ratios are acceptable if more tissue dispersion is desired.
- Pipette the media up and down multiple times with a 10 mL pipette to further disperse the DP in the media. Be careful to avoid large bubbles. Complete dispersion is nearly impossible due to the sticky nature of the tissue. However, it is also not necessary since cells will migrate outward from the tissue once plated.
- Transfer 1 mL of the dispersed DP to each well of a 24-well tissue culture plate (Figure 1F).
- Place the plate in an incubator at 37 °C and allow cells to attach and migrate out from the undispersed tissue for 48 h before changing media. Primary cells need to be plated at relatively high concentrations in order to reach 85-90% confluence within 1 week. If this is not be achieved after 1 week, discard the plate.
- Optional Genetic Manipulation of DP Cells
- To alter cell signaling pathways, harvest DP cells from genetic knockout mice or from mice in which a gene of interest is flanked by loxP sites. In the latter case, the gene can be deleted using Adenovirus-Cre-GFP (Ad-Cre-GFP) recombinase to remove the flanked gene, as described below. Use Adenovirus-eGFP (Ad-eGFP) as a control virus to ensure that the viral infection does not cause a cellular response.
NOTE: Ad-eGFP is controlled by the CMV promoter enhancer, which is very strong. Ad-Cre-GFP is regulated by an IRES, an internal regulatory region between the Cre and GFP, which is not very strong. This results in brighter fluorescence in Ad-eGFP cells than Ad-Cre-GFP cells. Confirm equivalent infection levels based on the total numbers of cells fluorescing, not on the levels of cellular fluorescence. - Prepare 500 μL of media containing virus and 10 μg/mL of polybrene per well. Polybrene assists with viral infection in cells near confluence10. This protocol is based on a multiplicity of infection (MOI) of 100 for Ad-eGFP and 200 Ad-Cre-GFP for efficient gene infection with little or no effect on cell viability. The estimated cell number is 4 x 104 cells/well of a 24-well plate:
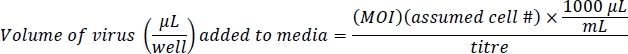
- Mix media with brief vortexing or pipetting and add 500 μL to each well.
- After 24 h, add an additional 500 μL of co-culture media that does not contain additional polybrene or virus.
- After a total of 48 h, aspirate virus-containing media and replace with fresh co-culture media. At this point, TG neurons can be added atop transwell filters.
- To alter cell signaling pathways, harvest DP cells from genetic knockout mice or from mice in which a gene of interest is flanked by loxP sites. In the latter case, the gene can be deleted using Adenovirus-Cre-GFP (Ad-Cre-GFP) recombinase to remove the flanked gene, as described below. Use Adenovirus-eGFP (Ad-eGFP) as a control virus to ensure that the viral infection does not cause a cellular response.
- Trigeminal neuron dissection, dispersion and plating
NOTE: In this protocol, the imaging of neurite outgrowth in co-culture with DP cells was optimized using adolescent (6-week-old) B6.Cg-Tg(Thy1–YFP)16Jrs/J mice. Central and peripheral nervous systems of Thy1-YFP mice have a yellow fluorescent protein (YFP) tag whose expression begins around P6-P10 in neurons and increases exponentially throughout the nervous system during postnatal and adult life11,12. YFP and GFP have conserved sequences that allow these nerves to be stained with anti-GFP antibodies, resulting in a pan-neuronal stain. Ultimately, these mice allow for better visualization and quantification of the neurons used and grown in cell culture.- Euthanize adolescent mice with carbon dioxide followed by cervical dislocation.
- Decapitate the mice and remove the skin from the skull. Be sure to include equivalent numbers of males and females.
- Insert the tip of a pair of micro-dissecting scissors into the base of the skull. Cut along the sagittal suture of the skull (Figure 1A).
- Make four small horizontal cuts: two along the coronal sutures by the ears, and two along the lambdoid sutures at the base of the skull. This should create two flaps of bone.
- Use the forceps to peel back the two flaps of bone. This should reveal the brain.
- Remove the brain. Transfer the head to a tissue culture dish with 1x PBS and put under the microscope.
- Locate the TG ganglia, which is easily visible in rodents13, housed in the dura matter between the brain and bone of the maxillary process (Figure 1B).
- Cut the three branches that travel to the eyes, maxillae and mandible and transfer the ganglia to cold 1x PBS using straight-edge fine forceps. Keep the dish containing the TG ganglia on ice during harvesting.
- Once all TG bundles are harvested, transfer ganglia to a 50 mL conical tube containing 5 mg/mL sterile-filtered collagenase type II using vial forceps.
- Vortex the collagenase with the TG bundle and place the tube in a 37 °C water bath for 25-30 min. During this time, take the conical tube out of the water bath, vortex, and return to the bath every 5-10 min.
- Centrifuge the collagenase-TG neuron solution for 2 min at 643 x g.
- Under a tissue culture hood, gently aspirate the collagenase with a micropipette.
- Add 5 mL of 1% sterile-filtered trypsin type II and vortex. Place the conical tube in a 37 °C water bath for 5 min.
- Centrifuge the trypsin-TG mix for 5 min at 643 x g. Remove the top portion of trypsin with a micropipette, so that the TG neurons are not removed. There will still be liquid in the tube.
- Add enough media to deactivate the remaining trypsin (at a 1:1 or lower ratio of trypsin to media).
- Count the number of cells and dilute the solution to 200,000 cells/mL (250 μL of cell-containing 50,000 cells).
- Place coated transwell filters from section 1.2 into wells with DP.
- Dilute the cell-containing solution so that there are 200,000 cells/mL. Pipette 250 μL onto the transwell filter, and culture the cells at 37 °C overnight (Figure 1F).
- The next day, replace the media with 1 mL of Co-culture media with 1 μM uridine and 15 μM 5'-fluor-2'deoxyuridine to stop the over-proliferation of mesenchymal cells that may prevent neurite outgrowth. Optional: Add growth factors or inhibitors in this media if attempting further manipulation.
- Culture the cells for additional desired time points. This protocol was optimized for 5 total days of culture with the media changed only on day 2 to add mitotic inhibitors. Longer time periods will require additional media changes.
3. Sample Collection and Processing
- Trigeminal staining
- Pipette 1 mL aliquots of sterile 1x PBS into a 24-well plate under a tissue culture hood for each transwell filter to be processed.
- Remove the liquid atop the filter be removed with a 200-1,000 μL pipette, being careful to leave cell layers intact. Attached cells should remain attached and unattached cells will come loose during this gentle pipetting. A vacuum filter can damage the cell layer and is not recommended for aspiration.
- Transfer the filter to the 1x PBS plate, being sure to remove the top layer of media as mentioned in the previous step.
- Place the plate with all transwell filters and lids on a rocker at 12 rpm or 40-50 rpm on an orbital shaker for 10 min.
- Aspirate the PBS, including the top layer as described above, add 500 μL of 1xPBS and place on the rocker or orbital shaker for an additional 5-10 min rinse.
- Aspirate the 1x PBS once more and replace with 4% paraformaldehyde (PFA). Use at least 500 μL so that the entire filter surface is submerged. Place the plate on a rocker at 12 rpm or orbital shaker at 40-50 rpm at room temperature for 1 h.
- Remove the PFA and rinse the plates twice for 5-10 min each on a rocker with 500 μL of 1x PBS.
- Block with 10% bovine serum albumin (BSA) + 5% goat/donkey serum, depending on the secondary antibody host animal, in 0.05% Tween-1xPBS (PBST). Use 450-500 μL of solution so that the filter is submerged in the liquid.
NOTE: At this stage, these plates can be stored at 4 °C for several months to process at a later time. Use parafilm to seal the plate to ensure evaporation does not occur and check regularly for evaporation so that that filter surfaces remain submerged. - Remove the block and add 450-500 μL of primary antibody in 1% BSA-PBST without an additional rinse. Incubate at 4 °C overnight, gently rocking.
- Remove primary antibody solution and rinse with 500 μL of 1xPBST on a rocker, 3 times, at room temperature
- Remove the PBST, add secondary antibody and incubate on a rocker overnight at 4 °C wrapped in aluminum foil to protect fluorophores from light degradation. Rinse again, 3 times, with 1x PBST on a rocker at room temperature. Replace with 1x PBS.
NOTE: At this point, the plate can be stored for several months at 4 °C if wrapped in aluminum foil to prevent fluorophore degradation. Use parafilm to seal the plate to ensure evaporation does not occur and check regularly for evaporation so that that filter surfaces remain submerged. It is best to image within one month. - For optimal imaging, utilize Thy1-YFP mouse neurons with an anti-GFP antibody to specifically stain the neuronal afferents well above background levels. Use Anti-Neurofilament 200 to precisely stain axonal structures if Thy1-YFP mice are not available.
- Image as desired. Filters do not need to be plated and can instead be placed atop a coverslip or slide for imaging with an inverted microscope.
NOTE: Areas of the filter will not contain afferent structures. Take several images with a z-stack depth that captures all afferent structures. Use stitching software to visualize the neurite outgrowth over large areas, or (preferably) entire filter regions.
- RNA, protein, and media collection
- While the transwell filters are processing, collect the media in RNAse/DNAse-free tubes and freeze for later assays (ELISA, proteomics, etc.).
- Immediately after media collection, aliquot lysis buffer or Radio Immuno Precipitation Assay (RIPA) buffer with proteinase and phosphatase inhibitors into the wells. This protocol was optimized for 100 μL/well in a 24-well plate.
- Allow buffers to lyse cells for 5 min, then scrape each filter with a new pipette tip, and collect the cell samples in RNAse/DNAse-free tubes. Freeze the lysis for future assays (semi-quantitative and/or quantitative PCR, Western blot, etc.).
- Optional immunofluorescence of dental pulp cells:
- Gently lift coverslips from Section 1.1 with forceps and transfer them to different wells with 4% PFA for 1 h with rocking.
- Aspirate PFA and rinse the coverslips twice with 1x PBS. Follow this with permeabilization, blocking and immunofluorescence for markers of interest with standard immunofluorescence techniques.
These results show that TG neurite outgrowth was increased in the presence of primary DP cells in the underlying well compared to the control of TG neurite monoculture (Figure 2A,C). There is some assay-to-assay variability in neurite outgrowth. Thus, a TG neuron monoculture should be included in all assays as a control to detect the basal levels of neurite outgrowth. Primary cells from the Tgfbr2f/f mouse were used in this protocol after infection of Ad-Cre-GFP and Ad-eGFP was confirmed in equivalent numbers of cells (Figure 2D). The Ad-eGFP served as a control viral vector. The Ad-Cre-GFP deleted the flanked gene, Tgfbr2, as demonstrated by semi-quantitative PCR (Figure 2E). In the cultures with Transforming growth factor beta receptor 2 (Tgfbr2) deletion, neurite outgrowth was decreased (Figure 2A-C).
We utilized the Thy1-YFP mouse TG neurons and stained them with an anti-GFP antibody that produced very specific and bright images of axonal structures well above this background, as shown in Figure 2. This allowed the specific staining of neuronal markers without non-specific staining of non-neuronal cells by utilizing previously reported methods such as crystal violet7. The large pores in the filters can autofluoresce and/or accumulate secondary antibodies and decrease the precision of axonal imaging (Figure 3). While the Thy1-YFP neurons with immunofluorescence drastically improves the imaging, further background can be removed with auto-thresholding software and then quantified. We also recommend performing immunofluorescence for Neurofilament 200 based on our preliminary findings (not shown) as well as others8,9 if Thy1-YFP mice are not available.
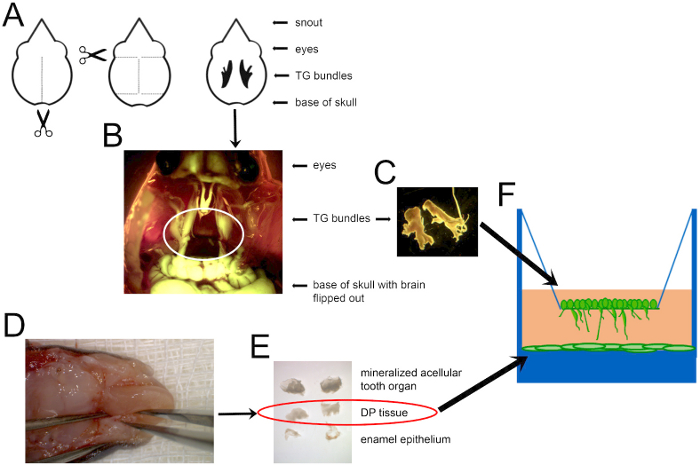
Figure 1: A schematic of the mouse dissection to obtain cells for co-culture. (A) A diagram of where to cut to open the mouse skull and locate TG nerves, shown in black in the last depiction. Scissors indicate where to insert the scissor tips to cut along the dotted lines. (B) A combined darkfield and GFP image showing Thy1-YFP+ TG nerves circled in white. (C) Dissected TG ganglia can then be dispersed and cultured, as shown in F. (D) The mandible of a P7 mouse, with forceps holding the mandible on the left and alveolar bone ridges containing unerupted teeth on each side of the tongue. (E) DP tissue (circled) extracted from the mineralized structure (top), and the enamel outer epithelium (bottom) that was removed to disperse and plate in a tissue culture-treated plate, as shown in F. Images are not shown to scale. DP cells were dispersed and grown to confluence before adding TG neurons. Please click here to view a larger version of this figure.
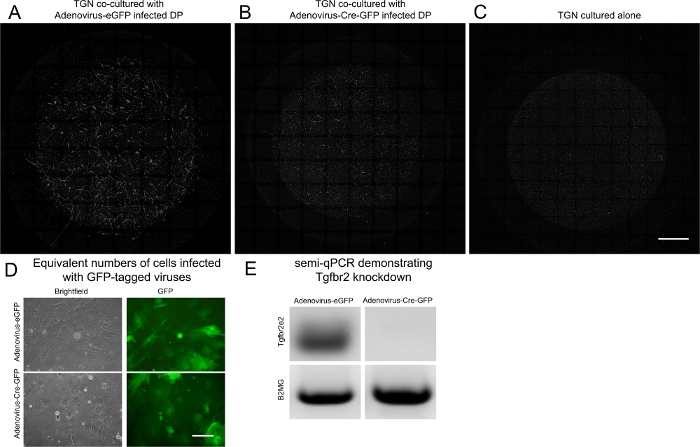
Figure 2: Representative results from co-culture. (A-C) Thy1-YFP TG neurons were cultured in transwell filters with 3 μm pores atop primary Tgfbr2f/f DP cells. Immunofluorescent staining was performed for the YFP protein using an anti-GFP antibody to provide highly specific staining of neuronal structures over the entire filter. The maximum projections of 100 μm z-stack confocal microscopy images at 10x were collected and stitched with stitching software. TG neurons demonstrated significantly more outgrowth when co-cultured with DP cells (A) than when cultured alone (C). Neurite outgrowth was not induced when neurons were co-cultured with DP cells infected with Ad-Cre-GFP to knock down Tgfbr2 (B). Scale bar = 1,000 μm. Equivalent numbers of cells infected with Ad-eGFP and Ad-Cre-GFP are shown in (D). Scale bar = 125 μm. Semi-quantitative PCR confirmed the Tgfbr2 KD (E). Please click here to view a larger version of this figure.
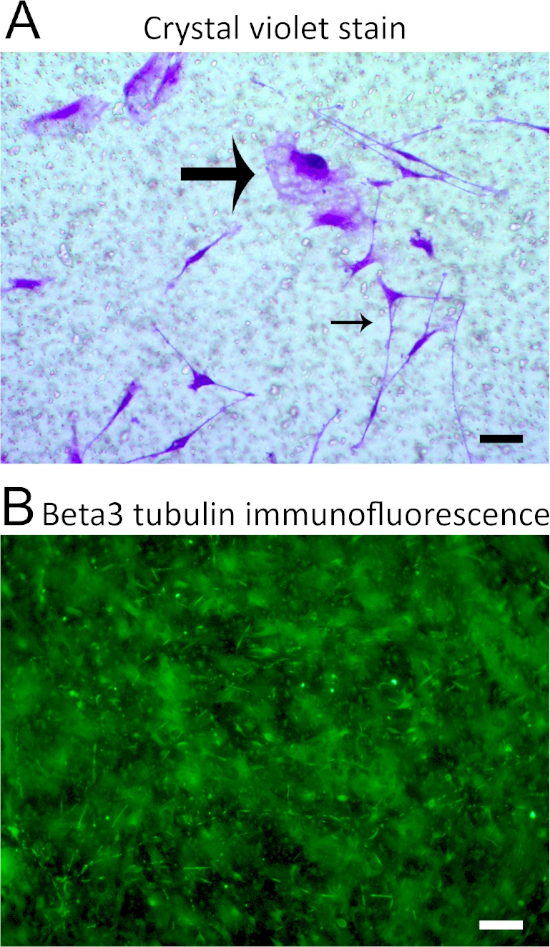
Figure 3: Technical difficulties presented in afferent imaging. (A) Brightfield imaging of transwell filters after crystal violet staining of cell populations. Large pores are prevalent. The large arrow points out a cell that exhibits mesenchymal morphology, whereas the small arrow points to a cell of neuronal morphology. Crystal violet stained both cells without bias. (B) Immunofluorescent staining of β3 tubulin with an Alexa-488 secondary antibody showed non-specific staining of multiple cells, making imaging of afferent structures difficult. Images are representative and were repeated over multiple assays to optimize the imaging shown in Figure 2. Scale bar = 50 μm. Please click here to view a larger version of this figure.
| Component | Volume | Concentration | |
| MEM α | 440 mL | ||
| Heat inactivated fetal bovine serume | 50 mL | 10% | |
| 100x L-glutamine | 5 mL | 1x | |
| Penicillin-streptomycin 100 x | 5 mL | 1x | |
| Change media on day 2 with mitotic inhibitors at these final concentrations | |||
| Uridine | 1 μM | ||
| 5'-Fluor-2'deoxyuridine | 15 μM | ||
Table 1: Co-culture media.
