Normal keratinocyte differentiation and RNA-seq analysis
In this experiment, keratinocyte lines derived from five individuals were used for differentiation and RNA-seq analyses. Figure 1 summarizes the experimental procedure of differentiation and RNA-seq analysis results. An overview of in vitro differentiation procedures of normal keratinocytes and cell morphology changes during differentiation are illustrated in Figure 1A. Principle component analysis (PCA) showed that keratinocytes undergoing differentiation had connected but distinct overall gene expression profiles (Figure 1B). Highly variable genes were clustered by kmeans to visualize gene expression dynamics and patterns during differentiation (Figure 1C).
Each cluster of genes was represented by the keratinocyte differentiation hallmark genes (e.g., KRT5 for proliferation, KRT1 and KRT10 for early and mid-differentiation, and IVL/LOR/FLG for late differentiation). Gene ontology (GO) annotation analysis of highly variable gene clusters (Figure 1C) showed a clear difference in gene functions of these gene clusters (e.g., keratinization in the mid-stages of differentiation; and epidermal cell differentiation, keratinocyte differentiation, and peptide cross-linking in the late stages; Figure 1D). Protein expression of several differentiation markers were measured by western blotting (Figure 1E).
P63 mutant keratinocyte differentiation and RNA-seq analysis:
In the second experiment, cell morphology and gene expression differences were compared between keratinocytes from healthy controls and three lines derived from patients carrying p63 mutations (mutants R204W, R279H, and R304W). Figure 2A shows an overview of differentiation procedures and cell morphology changes. Mutant keratinocytes remained flat on the surface of the dish and did not become crowded or overlap growth as control keratinocytes on day 7.
In PCA analysis, the control cell lines clearly followed a differentiation pattern, as compared to Figure 1B. However, the pattern of gene expression of mutant cells during differentiation stays largely similar to those of proliferating/undifferentiated cells. Among the three mutant lines, differentiating R279 samples moved along PC1 and PC2 to some extent, indicating that its differentiation was less impaired, compared to R204W and R304W.
In the clustering analysis (Figure 2C), genes downregulated in control cells (cluster 1) were partially downregulated in R204W and R279W, but their gene expression was not drastically changed in R304W. These genes likely play roles in cell proliferation, as shown by GO annotation (Figure 2D). In cluster 2 (Figure 2C), genes were first induced and subsequently downregulated in control cells. These genes are likely involved in keratinocyte differentiation, as epidermal differentiation and keratinization functions were highly enriched for this cluster genes (Figure 2D). These genes were not induced in R204W and R304W, whereas in R279H cells, these genes were induced but not downregulated as much as the control cells.
Genes in cluster 3 were only induced at the end of differentiation in control cells (Figure 2D). Consistent with this, these genes may have a role in the outer most layer of the epidermis, as they have been shown to be responsive to external stimuli and inflammation (Figure 2D). The expression pattern of these genes did not change much in all three mutant cell lines. The visible differences in gene expression patterns between control and mutant cells demonstrate that mutant cells cannot differentiate properly in these in vitro differentiation models.
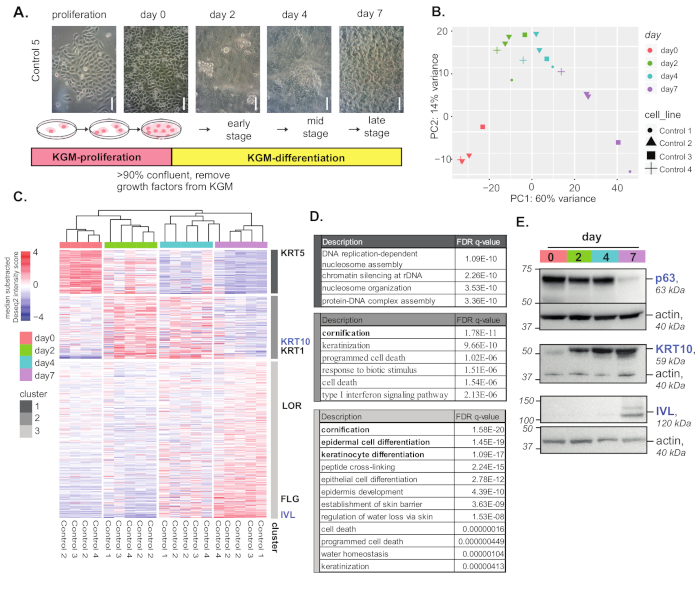
Figure 1: Keratinocyte differentiation and analysis. (A) Overview of the keratinocyte differentiation protocol and cell morphology. Scale bar = 100 µm. (B) Principle component analysis of differentiating control keratinocytes. (C) Heatmap of the top 500 highly variable genes during keratinocyte differentiation. Genes are clustered in three clusters using kmean clustering. Representative differentiation marker genes for each gene cluster are indicated at the side. (D) GO term enrichment analysis of overrepresented functions for genes in the highly variable gene clusters, as compared to a background of all expressed genes (counts of >10). GOrilla was used for the enrichment test. (E) Western blot of keratinocyte differentiation markers during differentiation. Please click here to view a larger version of this figure.
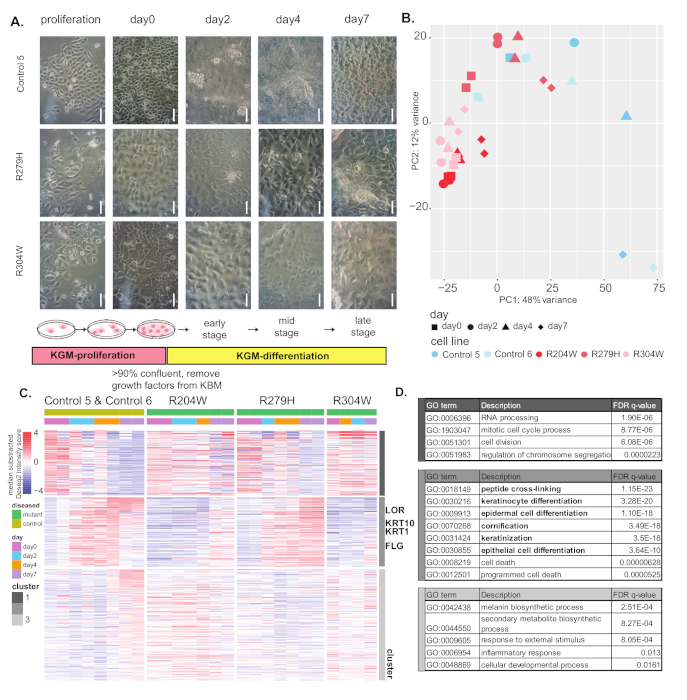
Figure 2: Comparison of control and p63 mutant keratinocytes. (A) Overview of the keratinocyte differentiation protocol and cell morphology of control and p63 mutant keratinocytes. Scale = 100 µm. (B) Principle component analysis of differentiating control and patient (R204W, R279H & R304W) keratinocytes. (C) Heatmap of differential genes between control and mutant keratinocytes. Genes are clustered in three clusters using kmean clustering. Representative differentiation marker genes for each gene cluster are indicated. (D) GO term enrichment analysis of overrepresented functions for genes in the highly variable gene clusters, as compared to a background of all expressed genes (counts of >10). GOrilla was used for the enrichment test. Please click here to view a larger version of this figure.
| KGM component | Stock | Medium | Volume |
| KBM | 500 mL | ||
| Pen/Strep | 100,000 units/mL | 100 units/mL | 5 mL |
| BPE | ~13 mg/mL | 0.4% | 2 mL |
| Ethanolamine | 0.1 M | 0.1 mM | 500 μL |
| o-phosphoethanolamine | 0.1 M | 0.1 mM | 500 μL |
| Hydrocortisone | 0.5 mg/mL | 0.5 µg/mL | 500 μL |
| Insulin | 5 mg/mL | 5 µg/mL | 500 μL |
| EGF | 10 μg/mL | 10 ng/mL | 500 μL |
Table 1. KGM-pro medium supplements.
| KGM component | Stock | Medium | Volume |
| KBM | 500 mL | ||
| Pen/Strep | 100,000 units/mL | 100 units/mL | 5 mL |
| Ethanolamine | 0.1 M | 0.1 mM | 500 μL |
| o-phosphoethanolamine | 0.1 M | 0.1 mM | 500 μL |
Table 2. KGM-diff medium supplements.
Supplemental Coding Files: Folder_structure.txt; Generate_genome.txt; Map_fastq.txt; RNA_seq_kc_differentiation_patient.html; RNA_seq_kc_differentiation_wt.html; Sample_data_example.csv; TrimGalore.txt; and Wig2bw.txt. Please click here to download this file.
