Targeting vector-based knock-in strategy
The hepatocyte nuclear factor 4 alpha (HNF4α) gene was chosen for targeted genome editing to introduce two point mutations into exon 8. A pair of Cas9n-sgRNA with 11 bp offset was designed close to the site to be modified. The piggyBac-based targeting vector worked as the homology-directed repair template for introducing the desired point mutations. A 967 bp 5'-HA and a 1142 bp 3'-HA incorporating synonymous nucleotide substitutions or the desired point mutations were amplified and cloned into the final targeting vector. The piggyBac insertion site was 16 bp and 22 bp away from the two desired point mutations. Colonies containing the puro-deltaTK selection cassette were selected with puromycin in the first round. Once the selection cassette was removed by transposase excision in the second round, the targeted site was modified seamlessly with only the desired point mutations incorporated in the gene (Figure 1).
Genetic editing in human pluripotent stem cells
To screen for correctly targeted cells, a three primer-based PCR method was used for genotyping (Figure 2). Sanger sequencing was performed to confirm the PCR results (Figure 3A). Post removal of the selection cassette, the modified region was sequenced again to confirm the correct introduction of desired point mutations (Figure 3B).
Colony establishment and characterization of edited human pluripotent stem cells
Colonies with the correct genotype were selected and expanded as needed. The established colonies need to be characterized before being used for further analysis. The edited cells possess the same morphology as the parental cells (Figure 4A). They also express representative human pluripotent stem cell markers, including transcription factors NANOG and OCT4 (Figure 4B), as well as cell surface markers SSEA4 and TRA-1-60 (Figure 4C).
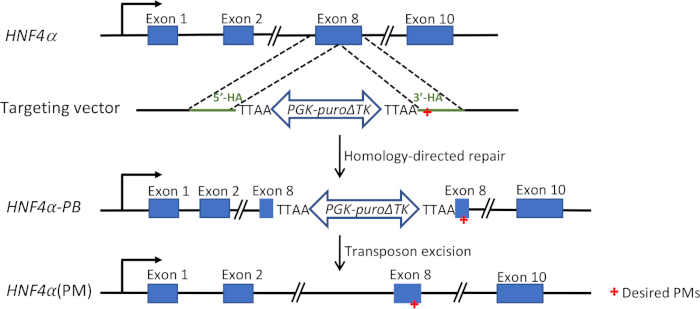
Figure 1: Targeting vector-based knock-in strategy11. A pair of Cas9n-sgRNA expression plasmids were used to induce DNA double-strand break in exon 8 of the HNF4α gene. A targeting vector with a selection cassette was used to introduce predetermined point mutations located 16 bp and 22 bp away. This selection cassette was contained within the piggyBac transposon, consisting of a positive-negative selection marker (puro-deltaTK) driven by a constitutively active promoter (PGK). Via homology-directed repair pathway, the targeted cells incorporated the selection cassette. Transposon excision mediated by transposase results in a seamless modification with only the point mutations present. Red cross indicates the location of desired point mutations. HA = homology arm; PB = piggyBac; PM = point mutation. Please click here to view a larger version of this figure.
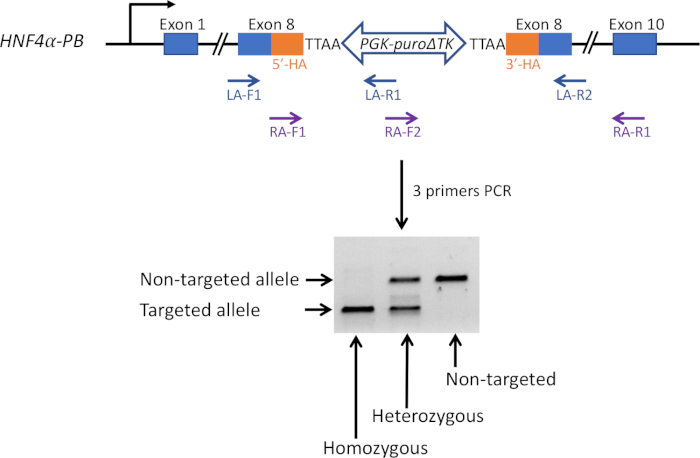
Figure 2: Three primer-based PCR method. To screen gene targeted cells, PCR-based genotyping was used. Three primers, LA-F1, -R1 and -R2 were used to amplify the left homology arm region. Independently, RA-F1, -F2 and -R1 were used to amplify the right homology arm region. Based on the gel electrophoresis result, non-targeted cells, and heterozygous and homozygous cells, were distinguished from each other. Please click here to view a larger version of this figure.
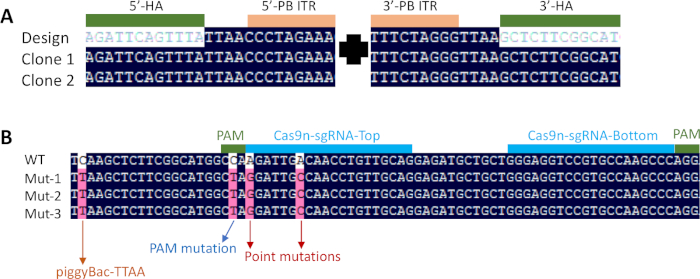
Figure 3: Sequencing results of genetically modified cells11. PCR products from two clones were sequenced and confirmed the correct insertion of the selection cassette at the targeted locus (A). 5' and 3' piggyBac inverted terminal repeats (ITR) were flanked by the TTAA direct repeats. Three clones were sequenced post transposon excision (B). A pair of Cas9n-sgRNA with 11 bp offset was used to introduce DNA double-strand break. Two predetermined point mutations (A to G and A to C) were introduced into the gene. One synonymous mutation was introduced to mutate the protospacer-adjacent motif (PAM), and another one to create the TTAA site necessary for piggyBac excision. Please click here to view a larger version of this figure.
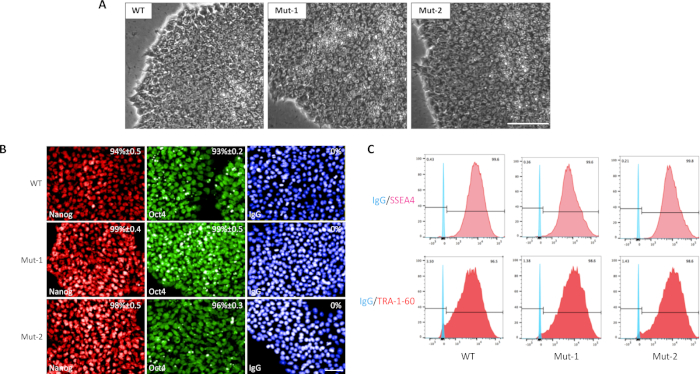
Figure 4: Characterization of edited human pluripotent stem cells. Morphology of two edited cell lines and the parental cells (A), scale bar = 100 µm. The expression of pluripotent stem cell markers NANOG and OCT4 examined by immunostaining (B, scale bar = 50 µm), in addition to SSEA4 and TRA-1-60 by flow cytometry (C) in two modified cell lines and the parental cells. IgG was used as a negative control. DAPI was used to stain the nucleus. The percentages were calculated as the average of three independent experiments. Please click here to view a larger version of this figure.
