A wildtype 10-week-old C57b6/j mouse heart typically results in between 75,000−150,000 atrial myocytes and 1.0−1.5 x 106 ventricular myocytes, equating to an approximate yield of 30%−50% for atrial and ventricular myocytes18,19. During and immediately after isolations, viable cardiac myocytes should appear rod-shaped and non-contracting. A majority of isolated cardiac myocytes should adapt this morphology, which is an indication of effective perfusion. The rod-shape morphology can also be a predictor of viability. The protocol aims to enhance the yield and viability of myocytes and non-myocytes isolated from a diseased mouse heart. Furthermore, it has been tested in a model of pressure overload-induced heart failure (data not shown).
To confirm adequate and replicable isolation of myocytes and non-myocytes from atrial and ventricular tissue, cells were observed and photographed at various days in culture (Figure 2). Additionally, quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed to measure the levels of transcripts that were cell type-specific. Cardiac muscle troponin T (Tnnt2) is a marker of cardiac myocytes and was robustly expressed in both atrial and ventricular cardiac myocyte cultures (Figure 3A). In contrast, atrial natriuretic peptide (Nppa, which is typically expressed exclusively in adult atrial cardiac myocytes under physiological conditions) and myosin light chain 2 (Myl2, which is a ventricular myocyte specific gene) were robustly and specifically expressed in atrial and ventricular cardiac myocyte cultures, respectively (Figure 3B,C).
Fibroblast markers, transcription factor 21 (Tcf21), platelet-derived growth factor receptor A (Pdgfra), and monocyte-derived cell marker cluster of differentiation 68 (Cd68) were exclusively expressed in non-myocyte cultures isolated from both atrial and ventricular chambers (Figure 3D−F). It is estimated that non-myocytes compromise ~65% of all heart cells and that a majority of these originate from a fibroblast or monocyte-derived lineage18,19,23,24. Thus, markers for these two lineages were chosen to be representative, given the interest in these cellular populations in studies of various models and etiologies of cardiac pathology.
Immunostaining of AMAMs and AMVMs for the t-tubule marker dihydropyridine (DHPR, which is a voltage-dependent (L)-type calcium channel) as well as the ryanodine receptor (RYR2) demonstrated intact t-tubules throughout isolation and long-term culture (Figure 4A,B). The abundance of DHPR and localization that was characteristic and unique to atrial and ventricular myocytes indicated the presences of t-tubules. Moreover, colocalization of DHPR with RYR2 immunostaining was an indicator of intact diad structures. Immunostaining for the sarcomeric protein alpha-actinin in atrial and ventricular cardiac myocytes resulted in the expected sarcomeric striation pattern. The sarcomeric striation pattern was used to assess the purity and viability of isolated cardiac myocytes in conjunction with rod-shaped morphological shape and nuclear staining with TOPRO-3 (Figure 4C,D; purple and red). As expected, ventricular cardiac myocytes were large, exhibiting an average length of ~150 mm, whereas atrial cardiac myocytes averaged ~75 mm. Furthermore, upon immunostaining analysis, atrial cardiac myocytes (but not ventricular cardiac myocytes) exhibited robust expression of atrial natriuretic peptide (ANP) in a staining pattern that was characteristic of localization to the endoplasmic reticulum and secretory granules (Figure 4C,D; green).
A characteristic unique to atrial cardiac myocytes is its classification as an endocrine cell in addition to contractile cell. While atrial myocytes secrete ANP under basal conditions, secretion increases in response to secretagogues (i.e., the alpha-adrenergic agonist, phenylephrine [PE]). Moreover, atrial cardiac myocytes secrete ANP and co-secretionally process a portion of the hormone from its precursor state (Pro-ANP, 15 kD) to the product peptide (ANP 3kD)16,17. This secretory ability can be quantified via immunoblot detection of ANP in the media of isolated atrial cardiac myocytes in response to acute PE treatment (Figure 4E). This secretory and processing ability of the atrial cardiac myocyte was found to be sensitive to culturing conditions. Thus, it is imperative that the atrial myocyte plating medium is supplemented with dexamethasone, insulin, transferrin, and selenium.
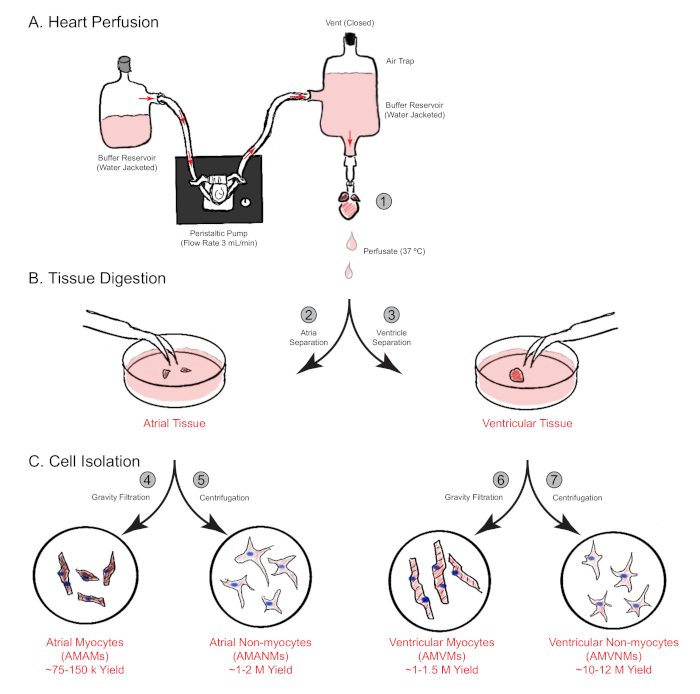
Figure 1: Schematic overview of retrograde heart perfusion, digestion, and cell isolation. Shown are the main steps involved in cell isolation from both atrial and ventricular chambers simultaneously from a single mouse heart. (A) A single mouse heart is rapidly cannulated via the ascending aorta and perfused in a retrograde manner. (B) The heart is separated into atrial and ventricular tissues for further digestion and physical separation. (C) Following adequate digestion, the cells are separated via gravity filtration into a total of four cellular fractions that are cultured for subsequent experimentation. Please click here to view a larger version of this figure.
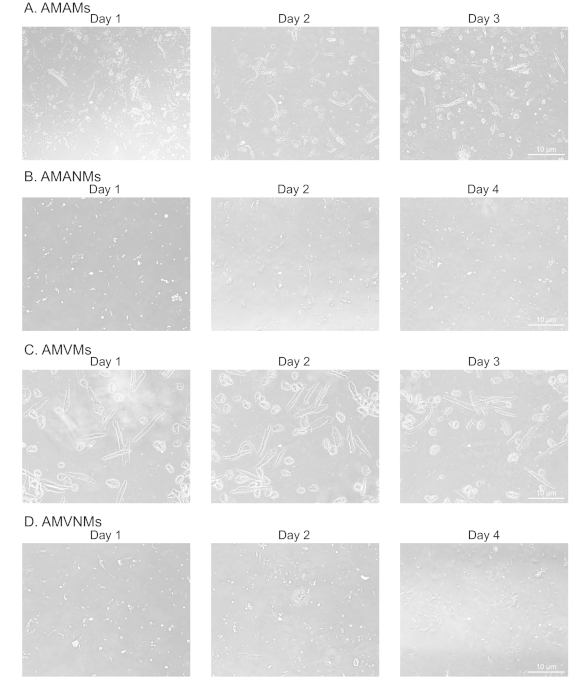
Figure 2: Morphological analysis of isolated atrial and ventricular cardiac myocytes and non-myocytes in culture. (A) Isolated adult mouse atrial myocytes (AMAMs), (B) adult mouse atrial non-myocytes (AMANMs), (C) adult mouse ventricular myocytes (AMVMs), or (D) adult mouse ventricular non-myocytes (AMVNMs) were plated at 5 x 105 cells/chamber on four-chamber (1.7 cm2) glass slides in respective plated media. Phase images were obtained at indicated days in culture using a 10x objective under an epifluorescence microscope. Please click here to view a larger version of this figure.
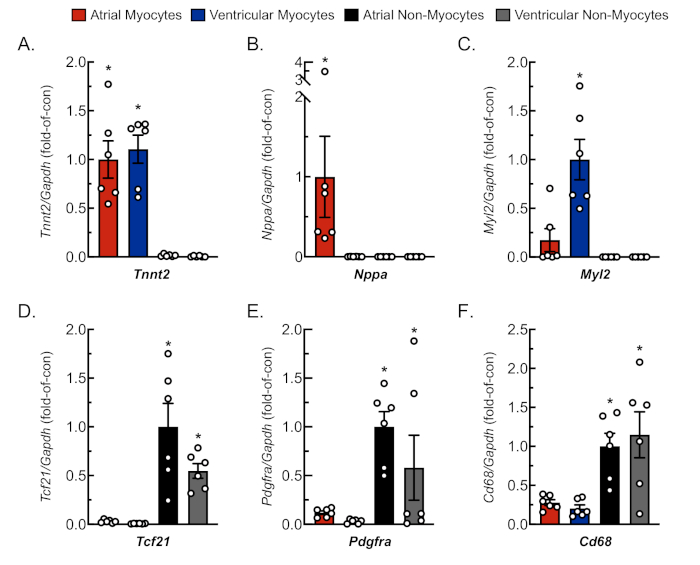
Figure 3: Representative qRT-PCR analysis of isolated cell cultures. RNA was extracted from freshly isolated cardiac myocytes and non-myocytes, and mRNA levels for cell-specific gene markers were determined by qRT-PCR4. (A) Tnnt2, cardiac muscle troponin T (cardiac myocyte marker); (B) Nppa, atrial natriuretic peptide (atrial myocyte marker); (C) Myl2, myosin light chain 2 (ventricular myocyte marker); (D) Tcf21, transcription factor 21 (fibroblast marker); (E) Pdgfra, platelet-derived growth factor receptor A (fibroblast marker); (F) Cd68, cluster of differentiation 68 (monocyte-derived cell marker). Data represent mean ± SEM (*p ≤ 0.05 different from all other values, as determined by ANOVA followed by Newman Keul’s post-hoc analysis). Please click here to view a larger version of this figure.
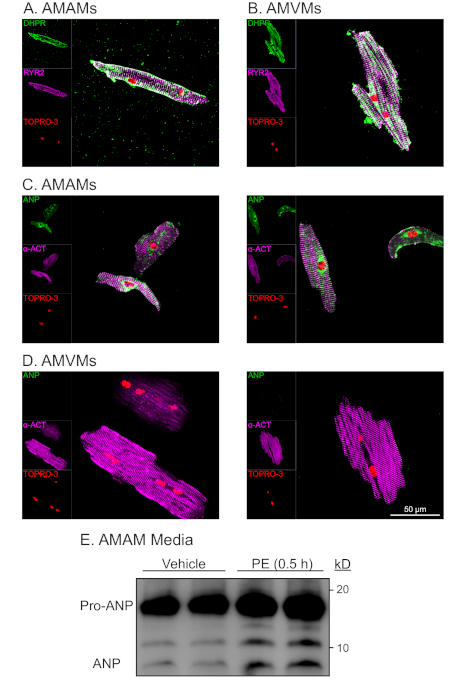
Figure 4: Representative morphological and functional analysis of isolated atrial and ventricular cardiac myoyctes. (A) AMAMs or (B) AMVMs were plated at 5 x 105 cells/chamber on four-chamber (1.7 cm2) glass slides in respective plating media for 1 h to allow for adhesion. This was followed by either refeeding atrial myocyte plating media or changing to ventricular myocyte maintaining media supplemented with blebbistatin for an additional 16 h. Cultures were subsequently fixed then immunostained for RYR2 (purple), DHPR (green), and nuclear stain TOPRO-3 (red). (C) AMAMs or (D) AMVMs were isolated and plated, then immunostained for a-actinin (purple), ANP (green), and TOPRO-3 (red). Shown are two representative images for each cell type. (E) AMAMs were plated at 5 x 105 cells/well on a 12 well culture dish for 16 h in atrial myocyte plating media. AMAMs were subsequently treated for 0.5 h with vehicle or the ANP secretagogue (phenylephrine, 50 mM) before media were collected and subjected to immunoblot analysis for ANP. Prior to immunoblot analysis, media samples were centrifuged at 500 x g for 5 min to remove cellular debris and ensure that the observed ANP was the result of active secretion from AMAMs. Please click here to view a larger version of this figure.
