This protocol (schematized in Figure 1) was used successfully to obtain C. elegans total protein extracts (Figure 2) for downstream immunoprecipitation of several proteins2 (Figure 3 and Figure 4). The presented bead mill homogenizer protocol was comparable in total protein extraction to dounce-based methods (Figure 2) and efficiently extracted nuclear (COL-19::GFP(NLS) (Figure 2) and cytoplasmic proteins (Figure 3 and Figure 4). Multiple samples of various sizes were extracted simultaneously (Figure 2). Argonaute proteins interact with members of the GW182 protein family, forming the miRISCs that bind to the target messenger RNAs and repress their expression10. Figure 3 shows successful co-immunoprecipitation of core miRISC components ALG-1 and AIN-1, consistent with previous reports11,17. More recently, efforts were made to identify additional protein interactors of Argonaute ALG-13 in order to learn more about how microRNA biogenesis and activity might be regulated by auxiliary factors. The RNA-binding protein HRPK-1 was identified in ALG-1 immunoprecipitates3. This interaction was recently confirmed in a reciprocal HRPK-1 immunoprecipitation experiment2. The presented extract and immunoprecipitation protocols successfully recovered ALG-1 in HRPK-1-specific co-immunoprecipitates (Figure 4). In addition, the ALG-1—AIN-1 interaction was tested in a variety of genetic backgrounds and HRPK-1 was shown to be unnecessary for the ALG-1/AIN-1 miRISC assembly2 (Figure 3). Supplemental figures are provided to show the full membrane probed (Supplemental Figure 1).
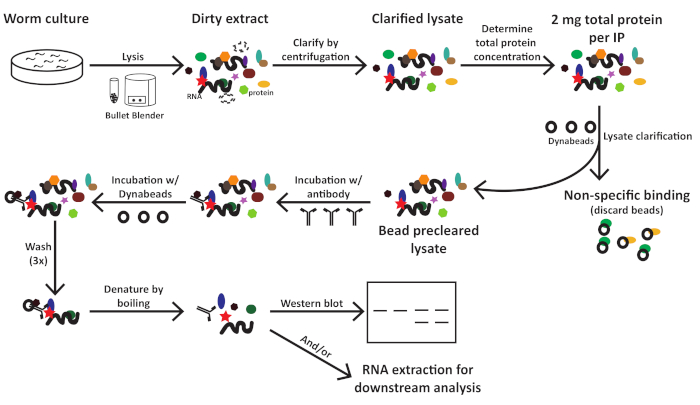
Figure 1: Workflow schematic for C. elegans extract preparation and immunoprecipitation. Please click here to view a larger version of this figure.
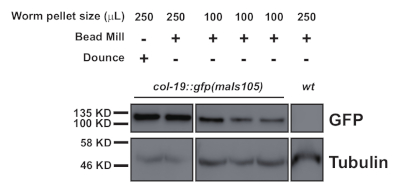
Figure 2: Western blot comparison of a nuclear localized GFP, COL-19::GFP(NLS), levels in dounce-prepared and homogenized samples from 250 µL and 100 µL worm pellets. Please click here to view a larger version of this figure.
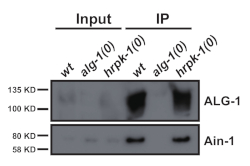
Figure 3: GW182 homolog AIN-1 co-immunoprecipitates with ALG-1. Western blotting for ALG-1 and AIN-1 proteins in ALG-1 immunoprecipitates. The ALG-1/AIN-1 co-immunoprecipitation was not affected by the absence of hrpk-1. Input = 10% of IP. Please click here to view a larger version of this figure.

Figure 4: ALG-1 co-immunoprecipitates with HRPK-1. Western blotting for HRPK-1 and ALG-1 in HRPK-1 immunoprecipitates is shown. Input = 10% of IP. * indicates antibody heavy chain. Please click here to view a larger version of this figure.
| M9 buffer (1 L) | |
| KH2PO4 | 3 g |
| Na2HPO4 | 6 g |
| NaCl | 5 g |
| 1 M MgSO4 | 1 mL |
| ddH2O | up to 1 L |
| 2x Lysis buffer (5 mL) | |
| HEPES (pH 7.4) | 200 µL |
| 2 M KCl | 250 µL |
| 10% TritonX | 100 µL |
| 1 M MgCl2 | 20 µL |
| 100% glycerol | 1 mL |
| ddH2O | up to 5 mL |
| Add fresh: | |
| 1 M DTT | 20 µL |
| EDTA-free protease inhibitor | 1 tablet |
| phosphatase inhibitor cocktail 2 | 100 µL |
| phosphatase inhibitor cocktail 3 | 100 µL |
| 1x Lysis buffer | |
| Dilute 2x Lysis buffer with an equal volume of ddH20. | |
| 1x Wash buffer 10 mL) | |
| HEPES (pH 7.4) | 300 µL |
| 2 M KCl | 500 µL |
| 10% TritonX | 100 µL |
| 1 M MgCl2 | 20 µL |
| 100% glycerol | 1 mL |
| ddH2O | up to 10 mL |
| 1 M DTT | 20 µL (add fresh) |
Table 1: Recipes
Supplemental Figure 1. Full probed Western blot membranes used to generate Figures 2-4 are shown. (A) Probed membrane for Figure 2. Note that membrane was cut to allow for simultaneous probing for GFP and Tubulin, reducing the overall blot size. (B) Probed membrane for Figure 3. (C) Probed membrane for Figure 3. *denotes antibody heavy chain. Please click here to view a larger version of this figure.
