1. Preparation of smFISH probes
NOTE: To label smFISH probes with a single fluorophore, follow a standard protocol for labeling nucleic acid oligonucleotides based on NHS ester chemistry21.
- Design smFISH probes. Decide whether to use “tiling” probes or “array” probes (Figure 1) for the gene of interest. See the Discussion section on how to make the decision.
- For “tiling” probes (Figure 1A), use an online probe designer tool (e.g., see Table of Materials).
- For “array” probes (Figure 1B), perform a BLAST sequence search to make sure that the probe sequence is not complementary to any other mRNA sequences.
- To study lacZ mRNA transcription and degradation kinetics, use two sets of 24 probes, each set covering the first and last 1 kb regions of lacZ (3,072 bp)19.
NOTE: These probe sets are, hereinafter, referred to as “5’ mRNA probe” and “3’ mRNA probe”, respectively. Sequences of these probes are listed in the Table of Materials.
- Order probe sequences as DNA oligonucleotides with a C6 amino linker at the 5’ end. Dissolve individual probes in water to 1 mM.
- Combine equimolar amounts of probes for “5’ mRNA probe” and “3’ mRNA probe” sets. For example, for the 5’ mRNA probe set for lacZ, combine 20 µL of each probe (total 24 kinds of probes in the set).
- Perform ethanol precipitation22 of the combined probes to remove any contaminations of primary and secondary amines (such as Tris, glycine, and ammonium salts) that can inhibit the conjugation reaction. In the end, dissolve the DNA pellet in 100 µL of water (yielding ~4.5 mM of DNA in a probe set).
NOTE: This step is recommended even if the probes underwent a standard desalt purification by the manufacturer. A standard filter-based purification may work in place of and in addition to the ethanol precipitation. - Choose two spectrally distinct fluorophores with a monofunctional NHS ester moiety, such that 5’ and 3’ mRNA probe sets can be labeled differentially. For example, prepare Cy5 NHS ester for 5’ mRNA probes and Cy3B NHS ester for 3’ mRNA probes. Dissolve each type of fluorophores in anhydrous DMSO to final 20 mg/mL (~25 mM).
- Prepare 0.1 M sodium bicarbonate (pH 8.5) right before each labeling reaction. Exposure to air for a long time will lower its pH and reduce the labeling efficiency.
- For the conjugation reaction, combine the following: 15 μL of the Cy5 fluorophore stock (from Step 1.5), 4 μL of 5’ mRNA probe set (from Step 1.4), 75 μL of sodium bicarbonate (from Step 1.6), and 7 μL of water. Wrap the tube with aluminum foil and shake at room temperature for 3-6 h.
NOTE: Longer incubation does not necessarily result in greater labeling efficiency. Also, the reaction can be scaled up or down if the concentrations of the components are maintained. - Repeat the above step for the 3’ mRNA probe set and the corresponding fluorophore (i.e., Cy3B NHS-ester).
- Perform ethanol precipitation22 to remove un-reacted dye molecules. Dissolve the pellet in ~50 μL of TE buffer (10mM Tris-HCl pH 8.0 with 1mM EDTA).
- Estimate the concentrations of DNA and fluorophore by using a UV-Vis spectrometer.
- Measure the absorbance at 260 nm and 559 nm (Cy3B) or 649 nm (Cy5). If the sample is too concentrated to yield an accurate measurement, dilute 1 µL of the sample to 10 µL.
- Convert the absorbance to the concentration:
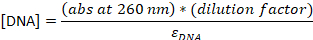
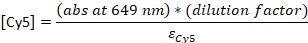
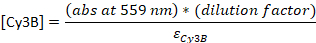
εDNA = 0.2 μM-1 (for 20-nt single-stranded DNA), εCy5 = 0.25 μM-1, and εCy3B = 0.13 μM-1
NOTE: [DNA] is the concentration of total probes within the solution. The concentration of individual probes is about 24x lower. The concentration of total probes will be used as “probe concentrations” from this point. If the ratio between [DNA] and [dye] is 1, the following HPLC step may be skipped23, and the sample should be diluted to final 4-5 μM in TE buffer.
- (Recommended) Purify the labeled probes from unlabeled probes and free dyes by using HPLC.
NOTE: Although this additional purification step will lead to the loss of sample, it is beneficial for the downstream applications. Removal of unlabeled DNA probes will increase the fluorescence signal from mRNA targets and removal of unreacted dyes will reduce background fluorescence.- Prepare HPLC with a standard analytical C18 column, 0.1 M triethylammonium acetate (TEAA) as buffer A, and acetonitrile as buffer B.
- Add 1 M TEAA to the sample (from Step 1.9) to make 0.1 M TEAA.
- Set the gradient program as follows: 0-5 min with 0% B, 5-35 min with a 0-30% linear gradient of B, 35-37 min with a 30-100% linear gradient of B, and 37-40 min with 0% B. Keep the flow rate at 0.1 mL/min and record chromatograms at 260 and 649 nm (for the Cy5-labeled samples) or at 260 and 559 nm (for the Cy3B-labeled samples).
- Collect the eluted sample when the absorbance increases in both DNA and fluorophore channels.
- Concentrate the eluted sample using a vacuum concentrator and re-suspend the pellet in 50-100 μL TE buffer.
- Check the concentration of DNA and fluorophore by using a UV-Vis spectrometer (see Step 1.10). Dilute, if necessary, to make final concentration around 4-5 µM. Store the probes at -20 °C
2. Preparation of solutions
- Prepare a large volume of DEPC-treated water and buffers (Table 1). These solutions can last over a year at room temperature.
- Prepare 4x fixing solution and wash solution (Table 1).
- Prepare the pre-hybridization solution and probe hybridization solution (Table 1). Prepare the probe hybridization solution during incubation in Step 5.1 or Step 6.1 and then keep the solution in a 37 °C countertop shaker for 20-40 min with a cover to minimize exposure to light.
NOTE: The concentrations of formamide, SSC, and probe were optimized for the lacZ probe sets to minimize background fluorescence while maximizing the real signal. See the Discussion section for details on how to modify these concentrations for different applications.
3. Preparation of coverslips and glass slides
- Clean coverslips and glass slides.
- Place individual coverslips and slides in a Coplin jar using forceps. Ensure that the coverslips and slides are separated and not touching each other.
- Fill the jar with 100% ethanol and close the lid. Place the jar in a water-bath ultrasonic cleaner and sonicate for 15-20 min.
NOTE: For the water-bath sonicator, it is recommended to turn off the heater function. - Pour out ethanol and wash with ultrapure water 3-4x. Use water flowing directly from the water purification machine.
- Pour out the water from the jar and fill it with 70% ethanol. Close the lid and perform sonication for 15-20 min and wash with ultrapure water.
- Fill the jar with ultrapure water and sonicate for 15-20 min.
NOTE: Coverslips and glass slides can be kept overnight in the Coplin jar filled with ultrapure water. - Take a slide or a coverslip out of the Coplin jar using clean forceps and blow-dry it using N2 gas. Repeat this for the remaining slides and coverslips.
- Place the dried slides in a clean storage box until use in Step 7.5. Place the dried coverslips in an empty 1,000-μL pipette tip box, which will serve as a “chamber” in the remaining procedure.
- Using a hydrophobic marker, draw circles on the coverslips following circular holes in the pipette tip box. These circles (~0.5 cm in diameter) will serve as “wells”. Wait at least 5-10 min for the marker to be completely dried.
NOTE: Always keep the lid of the tip box closed. - Apply a 20-μL drop of 0.1% poly-L-lysine to each well. Incubate for 10-50 min at room temperature.
NOTE: Adjust this volume according to the well size. Ensure that the solution completely covers the well area. For longer incubation, be careful to avoid evaporation. - After incubation aspirate poly-L-lysine without touching the surface as this will scrape the poly-L-lysine off. Then apply a drop (~20 μL) of DEPC water to the poly-L-lysine treated wells. Close the lid of the “chamber” to prevent evaporation until Step 5.1.
4. Time-course experiment and sample fixation
- Grow E. coli cells in ~20 mL liquid culture in a 250-mL flask. Keep the flask in a water bath shaker (30 °C) and continue shaking. Stop the shaker only when taking samples.
NOTE: Results presented in this paper are obtained from MG1655 cells grown in M9 minimal medium supplemented with 0.2% glycerol, 0.1% casamino acids, and 1 mg/L thiamine to an exponential growth phase (OD600~0.2). - Add 250 µL of the 4x fixing solution in an empty 1.5 mL tube. Repeat and prepare multiple tubes, as many as the time points to be taken. Label the tubes with time point numbers and keep them at room temperature.
- Take 750 µL of cell culture (OD600~0.2) before starting a time-course experiment. Add the culture to a tube marked for “time zero” (from Step 4.2). Invert the tube gently to mix cells with the fixing solution.
NOTE: Do not pipette up and down to mix, vortex, or “be rough” on the cells. This sample represents the repressed state and will be used as a control to calculate the fluorescence intensity of a single mRNA (see Step 9.4). - Add 0.02-1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) to the liquid culture to induce lacZ expression. Start a timer at this point (t = 0 min) and sample at a certain time interval (e.g., every 1 min) from then on. For sampling, repeat Step 4.3.
- Add 5 mM orthonitropheynl-β-D-fucopyranoside (ONPF) or 500 mM glucose24 at a certain time during the time-course experiment (e.g. at t = 1.5 min) to repress lacZ expression. After re-repression, continue to sample the cultures (Step 4.3) to track mRNA degradation.
NOTE: Repression can also be done with ~400 µg/mL rifampicin, a transcription initiation inhibitor25. - For fixation, incubate the tubes containing sampled cells at room temperature for 15 min, followed by incubation in ice for 30 min.
- To remove fixatives, centrifuge the tubes at 4,500 x g for 4 min at room temperature. Remove the supernatant with a pipette.
NOTE: Be sure to discard formaldehyde in a separate waste container following the safety protocol. - Add 1 mL DEPC-PBS and re-suspend the cells. Repeat centrifugation and re-suspension 2x more times.
NOTE: Fixed cells are fragile and need gentle treatment. Carefully re-suspend the pellet and avoid bubbles. - After the final wash step, re-suspend cells in ~30 µL DEPC-PBS.
5. Permeabilization of cell membranes
- Apply each time point sample to different wells on the coverslip (~30 μL per well). Wait for 10-30 min at room temperature for cells to adhere on the surface. Avoid merging of the liquid drops between wells.
- To rinse off unbound cells, aspirate the liquid and apply ~20 μL DEPC PBS to each well. Aspirate DEPC PBS within a few minutes.
- Permeabilize the cell membranes by applying 15 µL of 70% ethanol to each well for 4 min. Aspirate the ethanol after the 4 min, and make sure that the wells are completely dry.
NOTE: It is critical to limit the ethanol treatment for 4 min. Longer treatment will result in over-permeabilization. - Apply 30 µL of the wash solution to each well.
6. Probe hybridization
- Aspirate the wash solution from each well. Apply 30 µL of the pre-hybridization solution to each well. Incubate the chamber in the 37 °C oven for 30 min.
NOTE: Add ~50 mL of water to the bottom of the chamber to provide humidity. - Aspirate the pre-hybridization solution from each well. Apply ~30 µL of the probe hybridization solution to each well. Cover the chamber with aluminum foil and incubate in the 37 °C oven for 2 h.
NOTE: Make sure that the probe hybridization solution is in the 37 °C countertop shaker before this step. Avoid the merging of liquids between wells. Apply a smaller volume of the solution to each well, if needed.
7. Post-hybridization wash and preparation for imaging
- Using a multichannel pipette, apply ~30 µL of the wash solution to each well all at once. Aspirate and repeat 3-5x times of washing. Incubate the chamber in the 37 °C oven for 15-30 min.
- Repeat Step 7.1 two more times.
- Wash each well with DEPC-PBS 5x times. Follow the method used in Step 7.1 but skip the incubation process.
- Aspirate the liquid from the coverslip. Apply 4 µL of DEPC-PBS to each well.
- Using forceps, lift and flip the coverslip, and gently place it over a glass slide (from Step 3.2). Avoid bubbles.
- Seal the edges of the coverslip with silicone dental gum.
- Wait until the gum is solidified. One can pause here and store the slide overnight at 4 °C.
NOTE: Other smFISH protocols suggest adding oxygen scavenging reagents (e.g., glucose oxidase/catalase) or using a commercial anti-fade mounting medium14,26 to increase the photostability of the fluorophores.
8. Imaging
- To find an area of interest, use the live mode of phase contrast imaging. Change the field of view within a well by maneuvering the stage joystick. Choose an area where the cell density is optimal (i.e., there are many cells that are mostly separated). Adjust z-focus such that phase-contrast cell images are in focus.
- Take snapshots in the order of Cy5 (4-s exposure), Cy3 (2-s exposure), and phase contrast (0.2-s exposure).
NOTE: Cy3B dye molecules are imaged in the Cy3 channel, and the images are referred to as Cy3 images. - Repeat Steps 8.1-8.2 to acquire images of ~10 different areas within a well.
- Move the objective to another well and repeat Steps 8.1-8.3.
- Export images as TIFF files.
- (Optional) Image multi-color beads adsorbed on the coverslip surface in Cy5 and Cy3 channels to determine spatial shift between Cy5 and Cy3 channels for image registration purposes.
- Apply ~10 µL of multi-color fluorescent beads (0.2 µm diameter) on a clean coverslip surface and wait for 10-30 min. After washing with ~50 µL of PBS, apply ~5 µL of PBS and sandwich the coverslip with a glass slide. Seal and mount on the microscope.
- Image beads in both Cy5 and Cy3 channels.
9. Image analysis
NOTE: Matlab code used in this step is available in the following GitHub website: https://github.com/sjkimlab/Code_Publication/tree/master/JoVE_2020. The GitHub folder contains everything needed for the image analysis, including parameter values for cell segmentation and spot identification. The procedure in this step is further explained in the master script, called “FISHworkflow.m”.
- Open a cell segmentation tool, such as microbeTracker27 or Oufti28, and load phase contrast images. Choose “Independent frames” and press a button called “All frames” to begin the segmentation process, from which cells are identified and their contours are calculated (Figure 3B,C).
NOTE: Detailed protocols for using these software packages are available online (e.g., oufti.org). - Load Cy5 fluorescence images in the spotFinder function of microbeTracker or Oufti, and press the “Run” button to begin spot identification and quantification based on 2D Gaussian fitting (Figure 3B,C). Repeat this step for Cy3 fluorescence images to analyze spots in the Cy3 channel. This step produces a list of spots in each cell, including their intensities and coordinates.
- (Optional) Filter out dim spots (false positives) using a threshold, as explained in in the FISHworkflow.m file.
NOTE: Examine fluorescent spots in the negative control (e.g. MG1655 ΔlacZ) and determine the threshold to filter out false positives. - To obtain the spot intensity of a single mRNA, use a list of spot intensities measured at time zero (before adding IPTG), and fit the distribution of spot intensities with a Gaussian mixture model with two mixture components. Take the peak position of the first Gaussian population (black line in Figure 3D,E) as the spot intensity of a single mRNA. Perform this for Cy5 spots and Cy3 spots separately to obtain the spot intensity of a single 5’ and 3’ lacZ mRNA.
NOTE: Repeat this in every time-course experiment because the spot intensity of a single mRNA can vary slightly in different experiments. - Divide the fluorescence intensity of a spot with the intensity of a single mRNA (from Step 9.4) to obtain the number of mRNAs within a spot. Sum normalized spot intensities within a cell to calculate the total number of mRNA in a cell (Figure 3F). Perform these calculations for 5’ and 3’ mRNA separately.
- Calculate and plot the mean mRNA numbers per cell at each time point (e.g., Figure 4B), and analyze the in vivo kinetics of transcription and mRNA degradation from the temporal change in the mean mRNA levels (Figure 4B).
- To obtain the rate of transcription elongation, perform a least-squares fitting of a line to the initial rise in 5’ and 3’ mRNA signals and identify intercepts to the basal levels (Figure 4B). The difference between these intercepts indicates the average time for RNAPs to travel from the 5’ probe region to the 3’ probe region. Divide the distance between two probe sets (2 kb) with this time to obtain the average rate of transcription elongation.
- To obtain the rate of mRNA degradation, fit an exponential decay function, y = A·exp(-t/τ) to the final decay region of the 5’ and 3’ mRNA signals (e.g., Figure 4B). The fitting parameter, τ, is the average mRNA lifetime.
- (Optional) Analyze the cell-to-cell variation in gene expression (e.g., the cell-level response to the induction shown in Figure 4C), based on the distribution of mRNA numbers in each cell (calculated in Step 9.5).
- (Optional) Using information about spot location along the major and minor axes of a cell (obtained from Step 9.2), analyze the localization of mRNAs (Figure 4D,E).
- (Optional) Analyze co-localization of 5’ and 3’ mRNAs (Figure 5) by comparing localization of spots detected in the Cy5 and Cy3 channels.
- Load images of multi-color beads (Step 8.6) in the spotFinderF function in microbeTracker and obtain coordinates of bead centroids in Cy5 and Cy3 channels. Use the list of centroid coordinates to calculate the affine transformation matrix, which informs how Cy5 and Cy3 channels are shifted and rotated with respect to each other29.
- Apply the affine transformation matrix to Cy5 and Cy3 FISH images to convert Cy3 images in the Cy5 coordinate. Classify if a spot is co-localized with another spot in a different channel. For example, a spot in the Cy5 channel is considered to be co-localized with another spot in the Cy3 channel if the distance between their centroids is less than 150 nm (Figure 5).
- Analyze how many Cy5 spots are classified as “co-localized” with Cy3 spots at each time point. Also, analyze the intensity of the co-localized spots (Figure 5).
Figure 3 shows representative images from this smFISH protocol. A full field of view (86.7 μm x 66.0 μm using our microscopy setup detailed in Table of Materials) shows ~500 E. coli cells dispersed throughout the field (Figure 3A). If the density of cells is much higher than what is shown in this image, automatic cell segmentation becomes difficult as segmentation algorithms do not reliably identify individual cells when cells touch each other. One needs to adjust the concentration of cells and incubation time used for surface adherence (Step 5.1) to achieve the optimal density of cells in the field of view.
The morphology of cells in the phase contrast images should remain comparable to that of live cells for segmentation purposes (Figure 3A–C). If cells are over-permeabilized, the cell morphology changes (like “ghosts”; Supplementary Figure 1). In that case, one may reduce the duration of 70% ethanol treatment in Step 5.3.
Before induction the average lacZ expression level was ~0.03 mRNAs per cell, consistent with previous reports15,30. Also, the distribution of lacZ mRNA spot intensities before induction did not fit well with a normal distribution or a Poisson distribution due to the presence of spots with high intensities (Figure 3D,E). This suggests that most of the spots detected under the repressed state represent a single lacZ mRNA, but a small population of spots contains more than one lacZ mRNA. To isolate the population with a single lacZ mRNA, we used a Gaussian mixture model with two mixture components (insets in Figure 3D,E). Then, the mean of the first Gaussian was taken as the mean intensity of a single mRNA spot (e.g., the peak of the black curve in Figure 3D) and used to convert the spot intensity to the number of mRNAs, for any spots detected in the time-course experiment. To calculate the total number of mRNAs within a cell, the normalized spot intensities were summed in each cell (Figure 3F)19.
When the expression level of lacZ mRNA is low, there are one or two diffraction-limited lacZ mRNA spots spatially separated within a cell. Hence, the images of these spots can be analyzed by 2D Gaussian fitting for their intensity and localization.
When the expression level is high, such that spots overlap with each other within a cell, 2D Gaussian fitting does not do reliable quantification. In that case, the mRNA level should be calculated by dividing the total, background-subtracted fluorescence signal within a cell with the mean intensity of a single mRNA19.
When the expression of lacZ is induced, the signal of 5’ lacZ mRNA increases first and that of 3’ lacZ mRNA increases later (Figure 4B). If the expression of lacZ is repressed, both 5’ and 3’ lacZ mRNA signals decrease with some delay in between (Figure 4B). To obtain the rate of transcription elongation, the rise of 5’ and 3’ signals are first fit with lines (Figure 4B), and the difference in x-intercepts are taken as the time for RNAPs to travel the distance between two probe regions (2,000 nt). The rate of transcription elongation can be measured from each time-course experiment and standard deviations can be calculated from experimental duplicates. The average rate of transcription elongation was 15-30 nt/s under our experimental conditions19.
Additionally, the rate of mRNA degradation (inverse of the mean mRNA lifetime) was obtained by fitting the decay region with an exponential function (Figure 4B). Our time-course data contains mRNA degradation during and after transcription31. We fit the time points after 3’ mRNA started to decay (t > 6 min) to probe the degradation of released mRNAs. We obtained ~90 s as an average lifetime of either 5’ or 3’ lacZ mRNA19.
The rate of transcription initiation can be calculated from the slope of 5’ signal increase after induction (Figure 4B, blue), or from the average mRNA number at steady state (which is the initiation rate divided by the degradation rate). Furthermore, the probability of premature transcription termination can be estimated, either by taking the ratio between the slope of 3’ signal increase vs that of 5’ signal increase32 or between the steady-state levels of 3’ and 5’ mRNA regions19.
Because smFISH is a single-cell technique, we can analyze cell-to-cell variability in transcription. For example, one can analyze the percentage of cells expressing lacZ mRNA after IPTG is added (Figure 4C). One can also address whether mRNA localization changes after induction. We observed that 5’ and 3’ lacZ mRNA spots move slightly outward, away from the center of the cell (Figure 4D,E), consistent with a previous report33.
Lastly, analysis of co-localization between 5’ and 3’ mRNA spots can be informative (Figure 5A). For example, in the repressed state (time zero), about 25% of 5’ mRNA spots are co-localized with a 3’ mRNA spot. At t = 1 min, as many gene loci have 5’ mRNA synthesis, but not yet 3’ mRNA synthesis, most of the 5’ mRNA spots are by themselves without 3’ mRNA signal (i.e., low probability of co-localization). However, when the 3’ mRNA appears (i.e., t = 2 min), the probability of co-localization increases (purple arrow in Figure 5A,B). This time point, when the co-localization becomes frequent, depends on the rate of transcription elongation. The 2-D density plot of 5’ and 3’ lacZ mRNA numbers within each co-localization spot detected at this time point can be used to infer the density of RNAPs on the lacZ gene (Figure 5C). As previously reported19, the 5’ mRNA numbers in this plot indicate that most of the lacZ loci have less than 10 RNAPs on the DNA when lacZ expression is induced by 1 mM IPTG. Additionally, the 3’ mRNA numbers in this plot is related to the clustering of RNAPs34. The fact that the number of 3’ mRNA is close to one means that roughly only one RNAP enters the 3’ probe region. This suggests that RNAPs on the lacZ gene are spatially separated, instead of forming a cluster (or “convoy”).
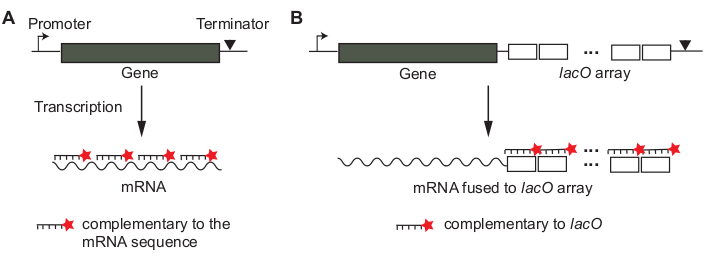
Figure 1: Design of smFISH probes for an mRNA of interest. (A) A tiling method. Sequences of short DNA oligonucleotides (~20 bp in length) are chosen so that they can cover the mRNA of interest. The oligonucleotide probes are labeled with a fluorescent dye molecule. (B) An array method. A non-coding array of tandem sequences (e.g., “lacO array”) is transcriptionally fused to the mRNA of interest. Fluorescently labeled probe complementary to the repeat unit (e.g., lacO probe of 17 bp in length) is used to amplify the signal of an mRNA. Please click here to view a larger version of this figure.
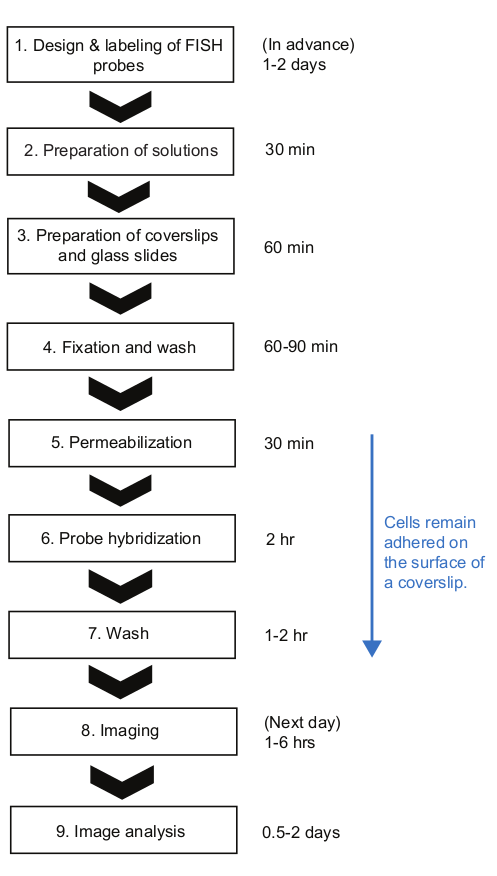
Figure 2: Schematic of smFISH experimental procedure and time duration of each step. Please click here to view a larger version of this figure.
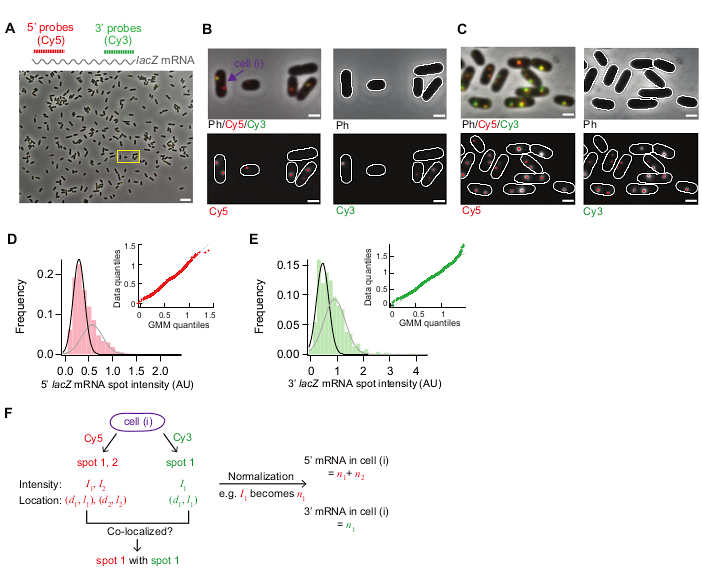
Figure 3: smFISH image analysis. (A–C) smFISH microscopy image of 5’ lacZ mRNA (red) and 3’ lacZ mRNA (green) in wild-type E. coli (MG1655) grown in M9 minimal medium supplemented with 0.2% glycerol, 0.1% casamino acids, and 1 mg/L thiamine at 30 °C. (A) A representative image of a sample from t = 3 min after induction with 0.05 mM IPTG at t = 0 min and repression with 500 mM glucose at t = 1.5 min. Phase contrast and two fluorescence images of Cy5 (for 5’ lacZ mRNA, red) and Cy3 (for 3’ lacZ mRNA, green) were overlaid with pseudo-coloring. The image shows an entire field of 86.7 μm x 66.0 μm. Scale bar, 5 μm. (B) Zoom-in version of a small region (yellow box) in (A). Cell outlines are shown in white, and fluorescence spots identified from image analysis are shown with red dots. Scale bar, 1 μm. (C) Detection of cell outlines and fluorescent spots under a high expression condition (t = 4 min after induction with 1 mM IPTG). Scale bar, 1 μm. (D–E) Distributions of 5’ and 3’ mRNA spot intensities measured before adding IPTG (the repressed state). The histograms are shown with two Gaussian functions (black and grey) whose mean values are from the Gaussian mixture model. Inset shows quantile-quantile plot of random numbers generated from the Gaussian mixture models and experimentally measured mRNA spot intensities (n = 1040 for 5’ mRNA and 680 for 3’ mRNA). (F) Information obtained for an individual cell pointed in panel (B). For a given cell (i), spots were identified in Cy5 and Cy3 channels, and their intensity (I) and coordinate along the short and long axis of a cell (d, l) were quantified from 2D Gaussian fitting. After normalization spot intensities were summed to yield the total number of 5’ or 3’ mRNAs in this cell. Also, co-localization between spots from different channels can be analyzed as in the example shown in Figure 5. Please click here to view a larger version of this figure.
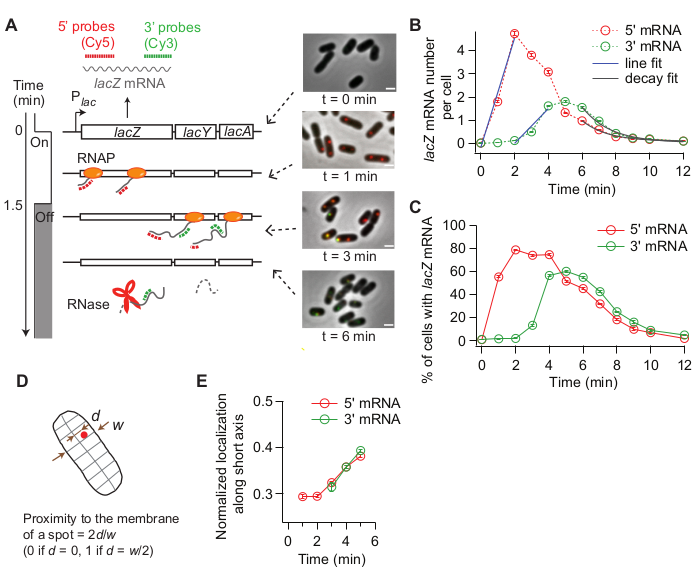
Figure 4: Analysis of in vivo kinetics of transcription and mRNA degradation. (A) Schematic and representative images of two-color smFISH experiments measuring changes in lacZ mRNA levels over time. Red and green dotted lines indicate Cy5 or Cy3B labeled oligonucleotide probes that hybridize to the 1-kb-long 5’ and 3’ mRNA regions of lacZ mRNA in E. coli, respectively. Also shown are overlays of two fluorescence images with a phase contrast image at indicated time points after induction with 0.2 mM IPTG at t = 0 min. Transcription was repressed with 500 mM glucose at t = 1.5 min. Scale bar, 1 μm. The figure has been modified from Kim et al19. (B) 5’ and 3’ lacZ mRNA numbers per cell over time, during the experiment described in the panel (A). Error bars are bootstrapped SEMs. At least 1,200 cells were analyzed per time point. The initial rise of the 5’ and 3’ mRNA signals was fit with a line (blue). The difference in x-intercepts was 1.93 min, yielding the average rate of transcription elongation of 17.3 nt/s. Final decay of the 5’ and 3’ mRNA signals was fit with an exponential decay function (grey). The fit parameters indicate that the average mRNA lifetime is 1.52 min for 5’ mRNA and 1.66 min for 3’ mRNA. (C) Percentage of cells with one or more lacZ mRNA spots during the experiment described in (A). Error bars are bootstrapped SEMs. (D) Localization of a spot along a cell’s short axis. One can quantify a spot’s proximity to the membrane by dividing the location along the short axis (d) with half width of the cell (w). (E) Change in the localization of 5’ and 3’ lacZ mRNA spots along cells’ short axis during the experiment described in (A). Please click here to view a larger version of this figure.
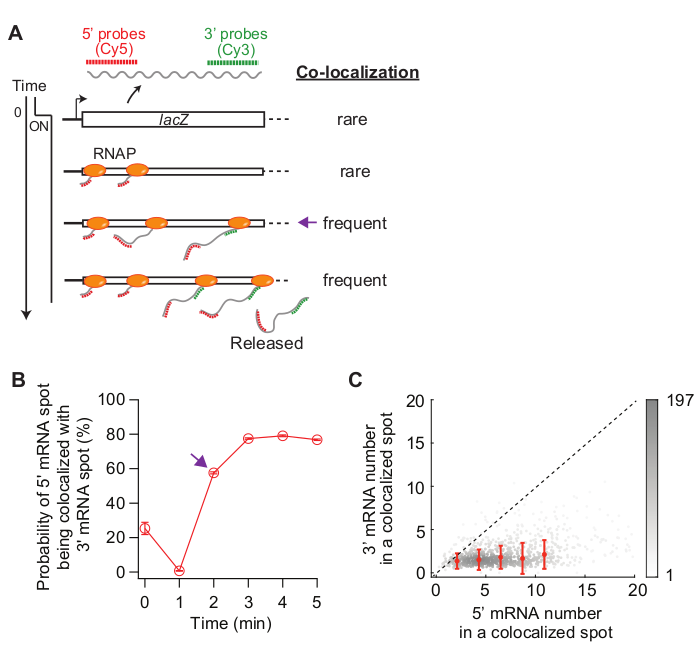
Figure 5: Analysis of co-localization of 5’ and 3’ mRNA spots. (A) Schematic showing the expected co-localization between 5’ and 3’ mRNA spots after induction. When 3’ mRNA is made, the probability of a 5’ mRNA spot being co-localized with a 3’ mRNA spot increases (purple arrow). (B) The probability of co-localization after induction with 1 mM IPTG. The purple arrow indicates the time point where the probability of co-localization first becomes frequent according to the schematic in panel (A). (C) The number of 5’ and 3’ lacZ mRNAs within a co-localization spot detected at t = 2 min after induction with 1 mM IPTG (total 841 spots). Gray dots represent individual co-localized spots, whereas red dots represent the average of binned data. Error bars are SEM. The shade of gray indicates the density of points in a given area of the graph. The dotted line indicates a slope of 1. Please click here to view a larger version of this figure.
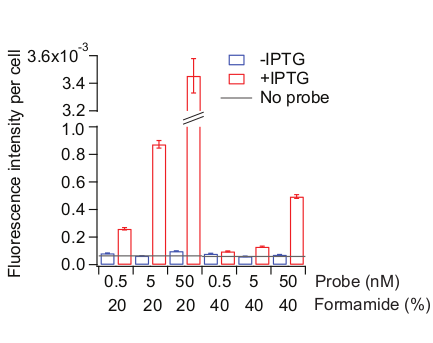
Figure 6: Optimization of the probe hybridization condition. Two kinds of samples were used: MG1655 cells grown as described in Figure 3 and remain uninduced (blue) or treated with 0.5 mM IPTG for 20 min (red). Probe hybridization solution was made with different concentrations of probes (total 72 Cy5-conjugated probes tiling the entire lacZ region) and of formamide. Formamide concentrations were also adjusted in the pre-hybridization solution and the wash solution, accordingly. “No probe” (grey line) indicates the fluorescence level of the IPTG-added cells treated with no probes during the hybridization step. Mean fluorescence intensity normalized by cell area (AU) was calculated from 300-800 cells. Error bars are bootstrapped SEMs. Please click here to view a larger version of this figure.
Supplementary Figure 1: Distorted cell morphologies due to over permeabilization. Overlay of phase contrast (gray scale), 5’ lacZ mRNA (Cy5, red), and 3’ lacZ mRNA (Cy3, green) images of MG1655 cells 5 min after the induction with 1 mM IPTG. (A) An example showing mixture of normal cells and overly permeabilized cells lacking normal morphology (indicated with pink arrows). (B) An example showing “ghosty” cells clumped together. Scale bar = 1 µm. Please click here to download this file.
| DEPC Water | ||||
| Add 0.1% DEPC to ultrapure water and incubate the bottle (covered) in the 37°C oven overnight and autoclave next day. | ||||
| DEPC PBS (10X) | ||||
| Mix the following: | ||||
| 80 g | NaCl (final 1.37 M) | |||
| 2 g | KCl (final 27 mM) | |||
| 14.2 g | Na2HPO4 (final 100 mM) | |||
| 2.7 g | KH2PO4 (final 20 mM) | |||
| Ultrapure water to 1L | ||||
| Filter (0.22 μm) into a glass bottle. | ||||
| Add 0.1% DEPC and follow the instruction for DEPC water. | ||||
| To make 1X solution, dilute 10 times with DEPC water. | ||||
| 1M DEPC sodium phosphate buffer, pH 7.4 | ||||
| Mix the following: | ||||
| 115 g | Na2HPO4 | |||
| 22.8 g | NaH2PO4 | |||
| Ultrapure water to 1 L | ||||
| Filter (0.22 μm) into a glass bottle. | ||||
| Add 0.1% DEPC and follow the instruction for DEPC water. | ||||
| 4X fixing solution (16% formaldehyde) | ||||
| 5 mL | 20% formaldehyde | |||
| 500 µL | DEPC water | |||
| 750 µL | 1M DEPC sodium phosphate buffer, pH 7.4 | |||
| Store at 4 °C for up to 2-4 weeks. | ||||
| CAUTION: Formaldehyde is toxic. Wear gloves and use a fume hood when making this solution. | ||||
| Wash solution | ||||
| Mix the following: | ||||
| 10 mL | Formamide (final 25%) | |||
| 4 mL | 20X SSC (final 2X) | |||
| Fill DEPC water to 40 mL | ||||
| Filter (0.22 μm) and store at 4 °C | ||||
| CAUTION: Formamide is toxic. Wear gloves and use a fume hood when making this solution. | ||||
| Pre-hybridization solution | ||||
| 200 µL | Formamide (final 20%) | |||
| 100 µL | 20X SSC (final 2X) | |||
| 10 µL | 100X VRC (final 1X) | |||
| 25 µL | 4% (w/v) BSA (final 0.1%) | |||
| 685 µL | DEPC water | |||
| NOTE: Vortex the VRC stock before taking 10 µL out. | ||||
| CAUTION: Formamide is toxic and a known teratogen. Wear gloves and handle it under a fume hood. | ||||
| Probe hybridization solution | ||||
| 200 µL | Formamide (final 20%) | |||
| 100 µL | 20X SSC (final 2X) | |||
| 10 µL | 100X VRC (final 1X) | |||
| 25 µL | 4% (w/v) BSA (final 0.1%) | |||
| 10 µL | 40 mg/mL E. coli tRNA (final 0.4 mg/mL) | |||
| 200 µL | 50% dextran sulfate (final 10%) | |||
| x µL | 5’ mRNA probe set (from Step 1.12) to final 4 nM. | |||
| y µL | 3’ mRNA probe set (from Step 1.12) to final 4 nM. | |||
| – | DEPC water to make the total volume 1 mL | |||
| NOTE: Add dextran sulfate last. Because it is very viscous, cut the end of a pipette tip before taking 200 μL out from the 50% stock. After adding dextran sulfate, pipette up and down to homogenize the solution. | ||||
Table 1: Recipes of the solutions used.
