In Sections 2.3 and 2.4, three examples of optimized crystal growth are presented, showing use of the instrument and an experimental design for growing large crystals. For this demonstration, we have used lysozyme as a model protein, although crystal growth experiments have been successfully performed with many other protein systems using this method (see above). By using and mastering the protocol presented here one can adapt it for other protein candidates.
In section 2.3 we demonstrated that established rational crystallization strategies could be beneficial in growing crystals with sufficient scattering volumes for neutron protein crystallography. Here, we demonstrate that the rational optimization strategies proposed also allow the generation of a uniform population of crystals of any specific size required for downstream structure determination approaches.
These two experiments are designed to emphasize the importance of phase diagrams in controlling crystal nucleation and growth. Here, control of the temperature and chemical composition of crystallization solutions in combination with monitoring the crystallization process in real time are used to study the qualitative phase diagram. Using this method, nucleation and crystal growth can be rationally optimized in a reversible manner. Use of such a serial approach also reduces the amount of protein and the time required to control the size and quality of the crystals.
In the dialysis method, a protein solution is separated from a crystallization solution by a semi-permeable membrane6 (Figure 5). This dialysis membrane allows small molecules such as additives, buffer, and ions to pass through the membrane but not macromolecules such as proteins6,20. This feature allows the crystallization solution to be modified during the course of the experiment6. Exchange of the solution can be done manually, for example in microdialysis buttons, or in an automated manner using an instrument developed for this purpose, OptiCrys8.
In the first set of experiments, microdialysis buttons were used for the crystallization of chicken egg-white lysozyme. Microdialysis buttons were immersed in crystallization solutions with different salt concentrations. In this simple crystallization grid experiment, the only variable is precipitant concentration whilst temperature is kept constant (293 K). As shown in Figure 4, slight variations in the salt concentration induce a change in the size and numbers of crystals observed, allowing investigation of the crystallization phase diagram. In Figure 4, panel 1, the crystallization solution contains 0.7 M NaCl and a limited number of larger crystals have appeared in the buttons. By increasing salt concentration from 0.7 to 1.2 M, supersaturation increases and the solution in the nucleation zone moves away from the metastable zone (Figure 4, panels 1 to 6). As a result, the number of crystals increases and their size decreases.
In the first experiment with a fully automated instrument enabling temperature-controlled dialysis crystallization, OptiCrys (Figure 9), the crystal growth experiment was tailored to generate large crystal growth. The experiment was launched at an initial temperature of 295 K with a crystallization solution containing 0.75 M NaCl and 0.1 M Na acetate buffer pH 4. Under these experimental conditions, the crystallization solution reached the nucleation zone in the vicinity of the metastable zone of the phase diagram (Figure 9, arrow 1). As a result, only a few nuclei were generated during the first stage of the experiment. In order to grow selected crystals further (shown in Figure 9), the crystal growth optimization workflow was driven towards the metastable zone by varying temperature as soon as the crystal-solution equilibrium was reached.
Each time equilibrium between crystal and solution was reached, the temperature was lowered, first to 291 K, then to 288 K and finally to 275 K, to keep the crystallization solution in the metastable zone. The result of this experiment is a single large crystal suitable for both macromolecular X-ray and neutron crystallography.
For most proteins, the precise quantitative phase diagram (or just a qualitative diagram) has not yet been obtained due to the lack of experimental devices capable of accurately measuring protein concentration (or just of observing/detecting the crystallization process in real time) during crystallization experiments18. As a result, it is often not possible to design the experiment in such a way that crystallization begins in the optimal area of the phase diagram, in the vicinity of the metastable zone.
Therefore, a crystallization optimization study must take place before the experiment dedicated to the growth of a large volume crystal is undertaken. In this study, using temperature variations (at constant chemical composition) on the one hand and variations in chemical composition (at constant temperature) on the other hand, are necessary to identify the metastable zone and to delineate the optimal conditions for starting a large crystal growth experiment.
To this end, two other experiments are presented which were tailored to demonstrate the reversibility of the temperature-controlled dialysis crystallization experiments with OptiCrys for nucleation, crystal growth, dissolution and re-growth. The crystal growth optimization workflow was controlled so that a uniform population of fewer, larger lysozyme crystals was grown, using variation of temperature or precipitant concentration.
In the second experiment with OptiCrys, the chemical composition of the crystallization solution was kept constant throughout the experiment (0.9 M NaCl in 0.1 M CH3COONa pH 4) with variable temperature. The initial temperature was set at 291 K. The results of this experiment are summarized in Figure 10. Because of high supersaturation, a large number of small crystals appeared in the crystallization chamber (Figure 10, panels 1 and 2). In accordance with the concept of direct protein solubility, by gradually increasing the temperature to 313 K, all of the crystals were dissolved (Figure 10, panels 3, 4 and 5). Finally, by lowering the temperature to 295 K, the second nucleation was initiated in the vicinity of the metastable zone and allowed controlled formation of a lower number of nuclei. Further crystal growth resulted in the uniform generation of a population of larger crystals (Figure 10, panel 7).
As shown in Figure 11, variation of the chemical composition of the crystallization solution, at a constant temperature of 291 K, can likewise be used to obtain a uniform population of larger crystals. Similar to the previous experiment, the initial condition was 0.9 M NaCl in 0.1 M CH3COONa pH 4. The NaCl concentration was then lowered gradually from 0.9 M to zero to dissolve the crystals (Figure 11, panels 4 and 5). At this point, NaCl was completely replaced by a buffer solution of 0.1 M CH3COONa pH 4. Reducing the salt concentration keeps the solution in the undersaturated zone of the phase diagram, which leads to the dissolution of the crystals. Then, a new crystallization solution with lower ionic strength, at 0.75 M NaCl in 0.1 M CH3COONa pH 4, was injected into the reservoir chamber. At this precipitant concentration, the first nuclei appeared (Figure 11, panel 6) after 90 minutes. The number of generated crystals was lower and the crystals reach a larger volume (Figure 11, panel 7) than before.
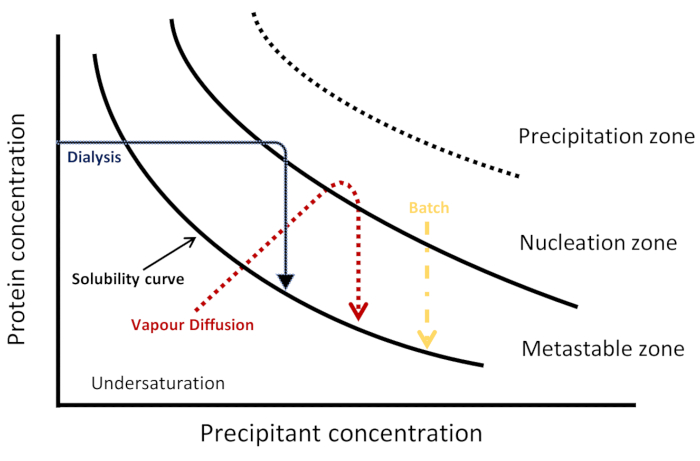
Figure 1: Schematic phase diagram. Kinetic trajectories for three crystallization techniques are represented in a salting-out regime. Each method achieves nucleation and crystallization differently, visualized by a different kinetic pathway through the phase diagram to reach the nucleation and metastable zones. The solubility curve separates undersaturation and supersaturation regions. Supersaturation is divided into three zones: metastable, nucleation and precipitation. In the nucleation zone, spontaneous nucleation occurs while in the metastable zone crystal growth takes place. This Figure is adapted from Junius et al.8 Please click here to view a larger version of this figure.
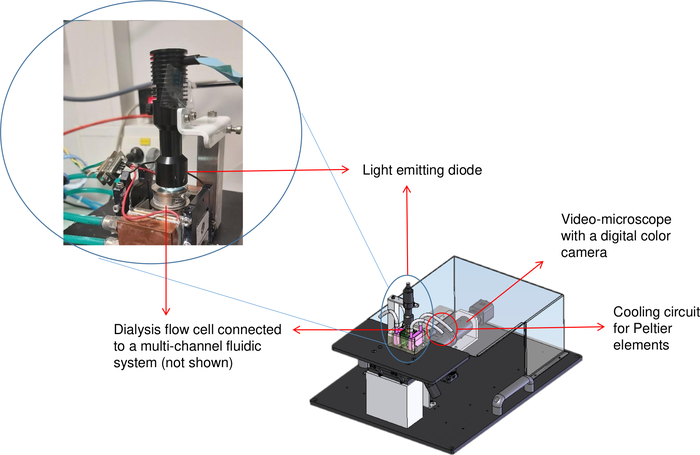
Figure 2: Schematic representation of the crystallization bench (OptiCrys). The LED light source is located on top of the temperature-controlled dialysis flow cell. An inverted microscope and the digital camera are shown at the top right of the image with the red arrow. The red circle represents the location of the chiller tubing. Please click here to view a larger version of this figure.
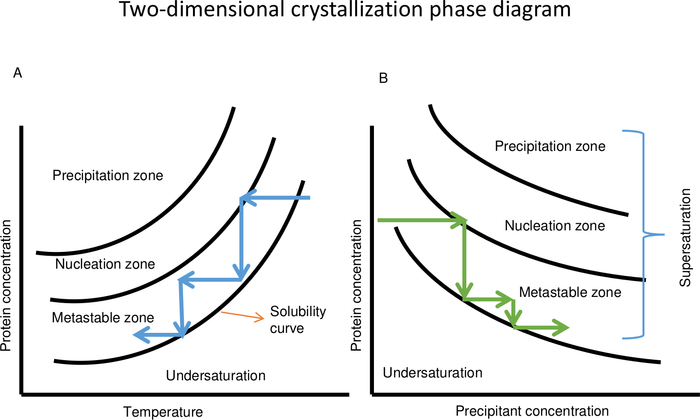
Figure 3: Schematic two-dimensional protein crystallization phase diagram as a function of temperature (A) and precipitant concentration (B). (A) In case of a protein with direct solubility, decreasing the temperature keeps the crystallization solution in the metastable zone. Temperature variation can be repeated several times to control the crystal growth process until crystals with the desired volume are obtained. (B) Changing the concentration of the precipitant solution can also be used to keep the crystallization solution in the metastable zone for growing crystals. This Figure is adapted from Junius et al.8 Please click here to view a larger version of this figure.

Figure 4: Crystals of lysozyme obtained using the dialysis method. This experiment was performed at a constant temperature of 293 K in 0.1 M sodium acetate buffer pH 4. Increasing NaCl concentration from 0.7 M to 1.2 M increases the nucleation rate and results in a larger number of crystals. Please click here to view a larger version of this figure.
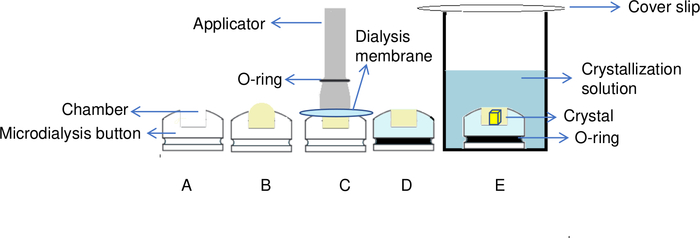
Figure 5: Overview of the protein crystallization process by the dialysis method. (A) By adding the protein to the chamber of the dialysis button, (B) a dome shape is created on the top of the chamber. (C) An applicator is used to transfer the O-ring to the groove of the dialysis button in order to fix the dialysis membrane in place. (D) The dialysis button is ready for immersion in the reservoir solution. (E) Crystallization solution passes through the semipermeable membrane and crystals start to form inside the chamber. Please click here to view a larger version of this figure.
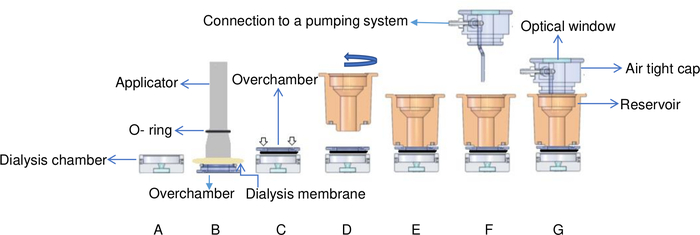
Figure 6: Schematic view of the temperature-controlled flowing dialysis setup. (A) The protein sample is added to the dialysis chamber. (B) The dialysis membrane is fixed onto the overchamber with an O-ring by using an applicator. (C) The overchamber is turned and fixed onto the top of the dialysis chamber. White arrows indicate where screws are placed on the overchamber. (D) The reservoir chamber is turned clockwise (E) and fixed on top of the overchamber. (F) The reservoir chamber is covered by an airtight cap with connectors to a pumping system and (G) the flow cell is placed in the brass support. This Figure is adapted from Junius et al.8 Please click here to view a larger version of this figure.
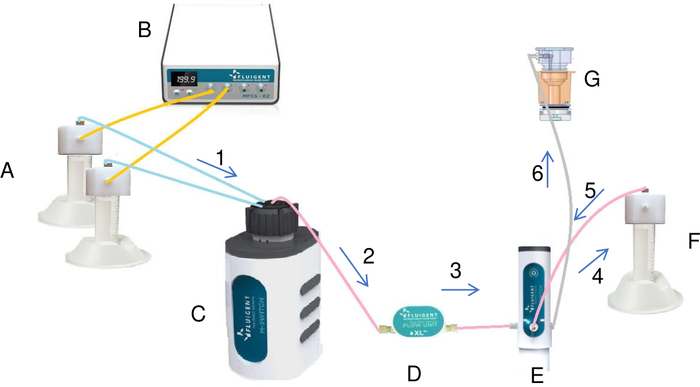
Figure 7: Preparation and injection of the crystallization solution in the reservoir by the fluidic system (A). Tubes containing salt and water are connected to the pressure/vacuum controller (B) and to the rotary valve (C). By using the pressure, pressure/vacuum controller creates a constant flow of the liquids from the tubes to the rotary valve. Each liquid passing through the flow meter (D) and the switch is injected into the mixing tube (F). Once all the liquids have been added to the mixing tube, the switch by some modifications injects the final solution from the mixing tube into the reservoir (G). The liquid flows through the system in the direction of the arrows in the diagram marked in ascending order (from 1 to 6). Please click here to view a larger version of this figure.
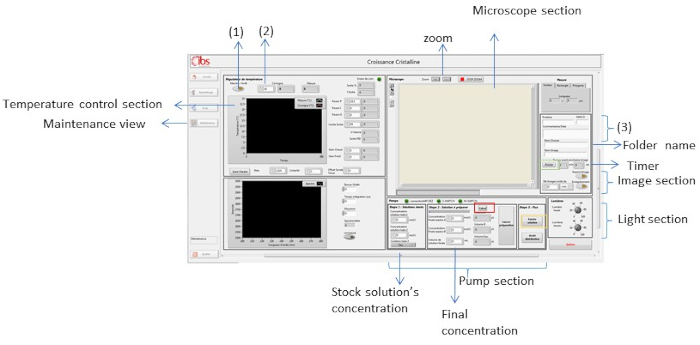
Figure 8: Maintenance view of the supervision software. This view is used to control different parameters like temperature, light, crystallization solution and zoom. Please click here to view a larger version of this figure.
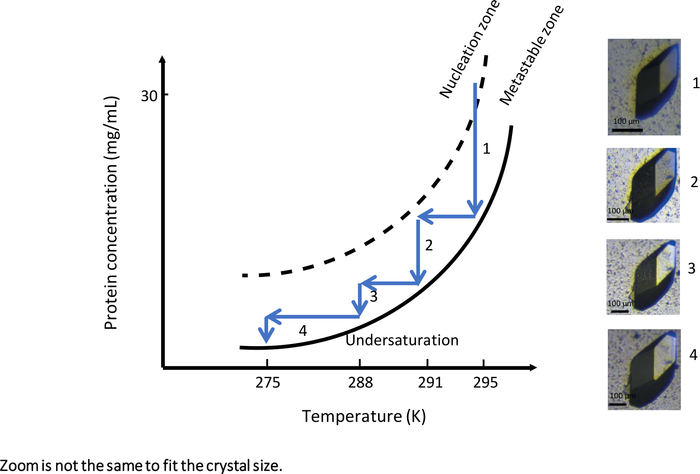
Figure 9: The phase diagram as a function of the temperature (selected images are to be tracked in ascending order). A single large lysozyme crystal is obtained by systematically changing the temperature from 295 K to 275 K. At each step, crystal growth is stopped upon reaching the solubility curve. Reducing the temperature by keeping the solution in the metastable zone restarts crystal growth. The images have different levels of magnification. This Figure is adapted from Junius et al.8,18 Please click here to view a larger version of this figure.
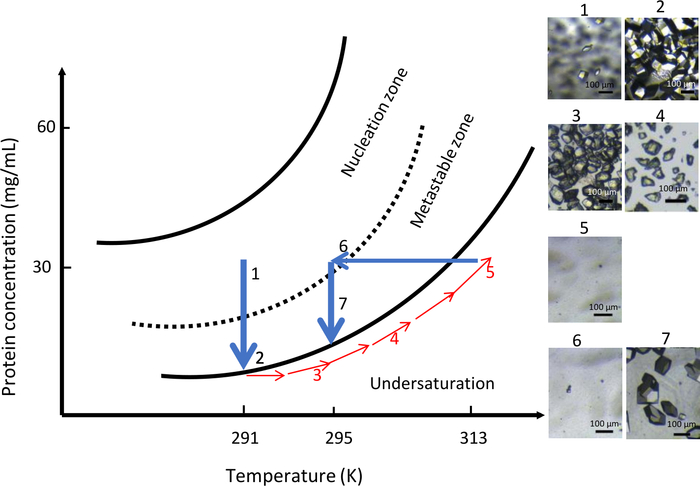
Figure 10: Optimization of crystal growth at constant chemical composition using temperature control (selected images are to be tracked in ascending order). Starting the nucleation process in the nucleation zone at 291 K, far from the metastable zone, results in the formation of numerous crystals. Increasing the temperature to 313 K then dissolves the crystals until no visible nuclei are seen in the dialysis chamber. Finally, decreasing the temperature to 295 K restarts the nucleation process for the second time leading to a limited number of larger crystals. This Figure is adapted from Junius et al.8,18 Please click here to view a larger version of this figure.
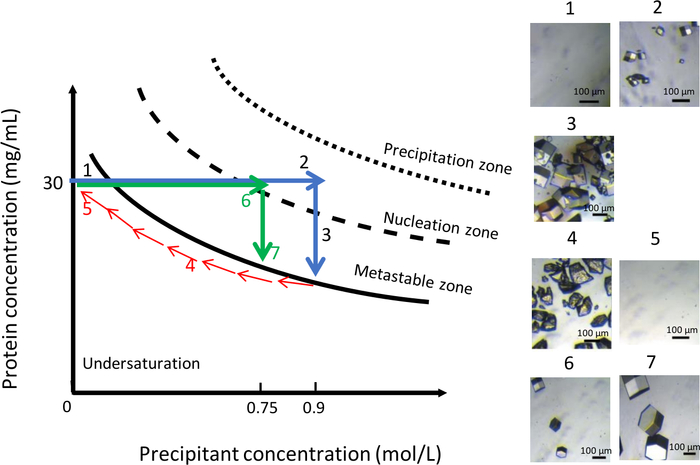
Figure 11: Optimization of crystal growth at constant temperature using variations in precipitant concentration (selected images are to be tracked in ascending order). Decreasing the precipitant concentration from 0.9 M to 0 M dissolves the crystals obtained during the first nucleation event. The crystallization process is restarted by the injection of the same precipitant but at lower ionic strength, 0.75 M, which leads to the formation of a few larger crystals. This Figure is adapted from Junius et al.8,18 Please click here to view a larger version of this figure.
