1. Preparation of reagents
NOTE: All steps in this protocol unless otherwise explained are carried out at room temperature.
- To prepare a solution of 1x PBS-BSA (0.1% w/v), dissolve 1 mg of Bovine Serum Albumin (BSA) in 1 mL of PBS. To prepare a stock solution of Thrombin 1,000 units·mL-1, dissolve 1,000 units of Thrombin in 1 mL of PBS-BSA (0.1% w/v). Make aliquots of 10 µL and store at -20 °C for up to 4 months.
- To prepare the culture medium for Drosophila brains, dissolve 1 g of Glucose in 1 L of Schneider’s medium in sterile conditions. Filter and store at 4 °C for up to 3 months.
- To prepare the Fibrinogen solution, dissolve 10 mg of Fibrinogen in 1 mL of culture medium for 20 min at room temperature or 5 min at 37 °C.
NOTE: If a different culture medium than the one prepared at step 1.2 is used and Fibrin clots already form at this step, the culture medium likely contains active Thrombin, which needs to be inactivated beforehand. A likely source of Thrombin is Fetal Bovine Serum, which can be inactivated by heating the serum to 56 °C for 30 min. - To prepare a coating solution of PBS-BSA (5% w/v), dissolve 50 mg of Bovine Serum Albumin (BSA) in 1 mL of 1x PBS.
- For the example reagent used in step 4, prepare a stock solution of 10 mM NAPP1 by dissolving 1 mg NAPP1 in 315 µL Dimethyl sulfoxide.
2. Dissection of Drosophila larval brains
NOTE: Perform this step under a dissection binocular.
- Use a brush to transfer L3 larvae into a 9-well borosilicate glass dish containing PBS. Stir to detach most of the fly food from the larvae and transfer to another well containing culture medium.
- Use forceps (e.g., Dumont #55) to grasp a larva across its entire diameter, in the middle of its body length (see Figure 1A,B). While holding the larva, transfer it to another well of the borosilicate plate containing 200 µL of fresh culture medium.
- Do not release the larva. To cut the larva in half across its entire diameter (Figure 1B), either grind the grasped area with lateral movement of the forceps tips (Figure 1A, top panel) or slide one tip of another pair of forceps between the two forcep tips holding the larva (Figure 1A, bottom panel). Do not cut the larva in half by pulling on one of its extremities, as it could damage the brain by pulling on it via the nerves crossing the body length (red lines in Figure 1B).
- Use forceps to hold the larva by its cuticle and another pair of forceps to peel apart the cuticle without pulling on the brain (Figure 1C). Repeat until the nerves originating from the brain are visible and the organs connecting the brain to the mouth parts can be accessed as shown in Figure 1D.
- Use forceps to hold the nerves originating from the brain. With the other pair of forceps, use either of the techniques shown in Figure 1A to cut the connection between the brain to the mouth parts, separating the brain from the rest of the cuticle (Figure 1D).
- Use forceps to hold the brain by the axons coming out of the ventral nerve cord. Pluck the organs depicted in gray in Figure 1E by gently pulling them away from the brain.
NOTE: Do not pull on the eye/antenna imaginal disks (dark yellow) to detach them from the brain. The connection between these tissues is too robust to be broken by pulling without damaging the brain. - While still grasping the nerves to hold and orient the brain, grasp the connection between the eye/antenna imaginal disks (dark yellow) and the brain with the other pair of forceps (Figure 1F). Use either of the techniques shown in Figure 1A to cut this connection without pulling on the brain. Check whether the brain is appropriately cleared of other tissues (Figure 1G) and not damaged (Figure 1H). Discard the brain if it shows signs of damage.
- Pipette the coating solution with a P20 in and out to coat the pipette tip. Pipette the brain with the coated tip along with 3 µL of medium (Figure 2A, left) and pipette it out in another well containing 200 µL of clean culture medium.
- Repeat steps 2.2–2.8 until enough brains are dissected, occasionally replacing the culture medium of the well in which the dissections are performed.
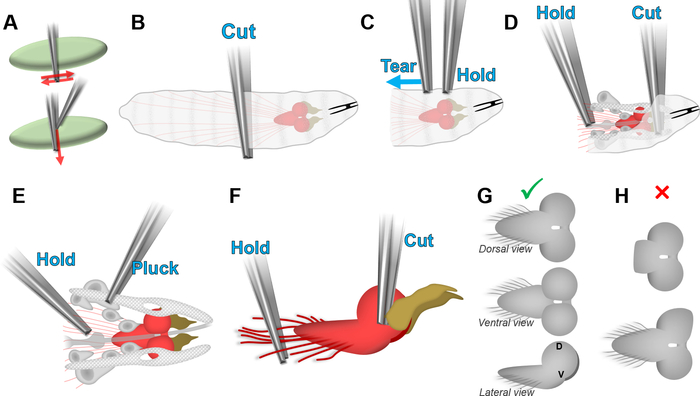
Figure 1: Dissection of D. melanogaster larval brains. (A) Technique for cutting without pulling using forceps. The sample (pale green) can be cut by being ground between the tips of the forceps (top panel) or by running a tip of a pair of forceps tip along the tips of a pair of forceps holding the sample (bottom panel). (B–F) Steps for isolating the larval brain without damaging it (see text). Red: brain. Dark yellow: eye/antenna disks. Blue text: actions to perform with the corresponding forceps. (G) Appearance of an isolated brain with no visible damage. D is the dorsal side and V is the ventral side on the lateral view. (H) Common damages caused by excessively pulling the brain during the dissection. Top panel: detachment of the ventral nerve cord. Bottom panel: deformation of a lobe. Posterior is left and anterior is right in all panels. Please click here to view a larger version of this figure.
3. Immobilization on coverslip with Fibrin clots
NOTE: Perform this step under a dissection binocular.
- Use a P20 pipette with a coated tip as in step 2.8 to transfer the dissected brains into another well of the borosilicate plate containing 200 µL of culture medium + Fibrinogen (Figure 2A).
- Pipette in one brain along with 9 µL of culture medium + Fibrinogen. With the end of the tip almost touching the cover glass bottom of a 35 mm cell culture dish, pipette out the contents (the culture medium and the brain) making the culture medium touch the coverslip immediately after exiting the pipette tip so that the brain and the medium do not slide to the sides of the tip, potentially damaging the brain (Figure 2A).
- Use one tip of a pair of forceps or a closed pair of forceps to gently push and position the brain within the culture medium + Fibrinogen drop.
NOTE: Touching the drop with an open pair of forceps can result in the culture medium getting pulled by capillarity between the forceps tips (Figure 2B). - If the ventral part of the brain has to be imaged, induce Fibrinogen clotting as follows to properly orient the brain within the clot (Figure 2C).
- Push the brain to one side of the drop. With a P1 pipette equipped with a P1 tip, pipette 1 µL of Thrombin solution, touch the edge of the drop on the opposite side of the brain with the pipette tip, and pipette out the Thrombin (Figure 2C, first drawing).
- Wait for 1–2 min for the Fibrinogen to start polymerizing, resulting in the formation of a cloudy precipitate at one side of the drop (Figure 2C, second drawing). Gently push and “tuck” the brain into the Fibrin clot, making sure that the part to image (e.g., the ventral side) is as close as possible to the coverslip without deforming the brain (Figure 2C, third drawing).
- Pipette out 1 µL of Thrombin solution close to the side of the brain not tucked into the Fibrin clot. (Figure 2C, fourth drawing).
- Wait for 2–3 min for the second Fibrin clot to set (Figure 2C, fifth drawing). Press on the edges of the clot to make it adheres more strongly to the coverslip, taking care not to deform the brain (Figure 2C, sixth and seventh drawings). If the brain appears to be too far from the coverslip, bring it closer by pressing the Fibrin clot close to the brain.
- If the dorsal part of the brain must be imaged, induce Fibrin clotting as follows (Figure 2D).
- Position the brain in the center of the drop, dorsal part facing the coverslip. With a P10 pipette equipped with a thin tip, pipette 1 µL of Thrombin solution, touch the edge of the drop on the opposite side of the brain with the pipette tip, and pipette out the Thrombin (Figure 2D, first drawing).
- Wait for 2–3 min for the Fibrinogen to start polymerizing, resulting in the formation of a cloudy, fibrous precipitate (Figure 2D, second drawing). Press on the edges of the clot to make it adhere more strongly to the coverslip, taking care not to deform the brain (Figure 2D, third and fourth drawings).
- Repeat steps 3.1–3.5 if several clot samples have to be immobilized on the coverslip.
- With the end of the tip positioned about 0.5 cm above the clot(s), gently pipette 390 µL of culture medium without Fibrinogen dropwise on top of the clot(s) (Figure 2B, right Petri dish). Do not add the culture medium on the sides of the clot as it may detach it from the coverslip.
- To wash out the remaining Thrombin, use a pipette to remove 300 µl of the culture medium and add 300 µl of clean culture medium, making sure that the clots remain fully immersed. Repeat twice to wash out excess Thrombin.
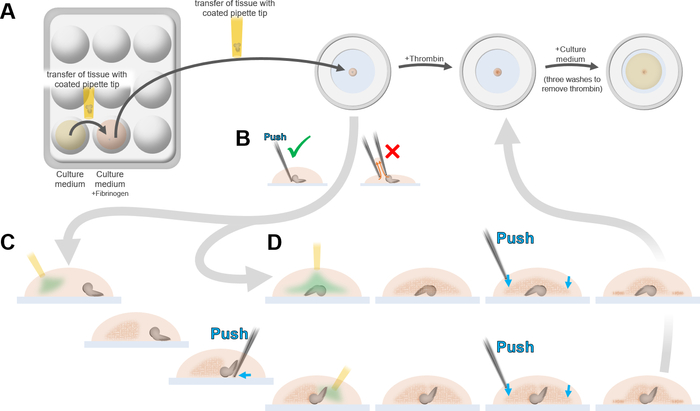
Figure 2: Immobilization of a Drosophila larval brain using Fibrin clots. (A) Using a BSA-coated Pipette tip, brains are transferred from the culture medium (yellow) into a culture medium + Fibrinogen solution (orange), then pipetted onto the coverslip (light blue) of a culture dish along with 3.5 µL of culture medium + Fibrinogen. Fibrinogen clotting is induced by addition of Thrombin to immobilize brains in dorsal or ventral position (see D and E). The secured brains in the clots are subsequently covered in culture medium without Fibrinogen and excess thrombin is removed by adding and removing culture medium three times. (B) The position of the brain within the drop of culture medium + Fibrinogen (orange) on the coverslip should only be adjusted by gently pushing it with the tip of a closed pair of forceps, or only one forceps tip. Touching the drop with on open pair of forceps can result in the culture medium getting pulled by capillarity between the tips of the forceps. (C) Steps for positioning the larval brain for imaging the ventral side (see text). (D) Steps for positioning the larval brain for imaging the dorsal side (see text). In (D) and (E), green: Thrombin. Dark orange: Fibrin clot. Pale blue: coverslip. Blue arrows: edges of the clot to be pushed against the coverslip to stabilize the clot. Please click here to view a larger version of this figure.
4. Addition of reagents during live imaging
- To avoid temperature changes causing changes of focus, keep the solution with the reagents to be added to the culture dish at the same temperature as the culture dish itself to bring it to the same temperature before adding it. If an environmental chamber is being used, leave the solution within the chamber.
- Remove the lid of the culture dish without displacing the culture dish itself. For easier removal, place the lid of the 35 mm dish upside-down on top of the dish before imaging.
- With a P1000 pipette, position the pipette tip close to the surface of the culture medium inside the culture dish and gently release the adequate volume of reagent solution onto it to reach the desired reagent concentration (e.g., 400 µL of a solution of 20 µM NAPP1 onto 400 µL of culture medium for a final concentration of 10 µM NAPP1), without generating strong fluxes that could detach the clots.
- To homogenize the solution within the culture dish, use a P200 pipette to slowly pipette a small amount of the solution in and out five times (e.g., 150 µL for a volume of 800 µL within the dish).
- Replace the lid on top of the culture dish without displacing the culture dish itself and resume imaging.
5. Washout of reagents during live imaging
- Prior to the washout, keep the washing solution at the same temperature as the culture dish.
- Remove the lid of the culture dish without displacing the culture dish itself.
- With a P200 or P1000 pipette, slowly remove some culture medium from the culture dish without exposing the top of the clot (e.g., remove 600 µL from 800 µL of culture medium within the dish).
- To dilute the reagent, with a P1000 pipette, position the pipette tip close to the surface of the culture medium inside the culture dish and slowly release the washing solution (e.g., add 1 mL of washing solution to the 200 µL of culture medium left within the dish for a dilution factor of 6).
- Repeat steps 5.3 and 5.4 until the reagent is diluted as required (e.g., another three similar rounds will result in a dilution factor of 1296, reducing the initial concentration of 10 µM NAPP1 to 7.7 nM).
- Replace the lid on top of the culture dish without displacing the culture dish itself and resume imaging.
6. Correction of the xyz drifting on tridimensional and/or multichannel time-lapses
- Preparation
- Download and install ImageJ24. Download the TurboReg25 and MultiStackReg26 plugins and place them inside the plugins folder of ImageJ.
- Download the MultiHyperStackReg (Supplemental coding file 1) and AutoHyperStackReg ImageJ macros (Supplemental coding file 2). To install a macro, either place the .ijm file inside the plugins folder of ImageJ or add the contents of the .ijm file to the ImageJ/macros/StartupMacros.txt file.
- On ImageJ, open a time-lapse movie including several slices and/or several channels (henceforth referred to as “hyperstack”, Figure 3A,4A). If the time-lapse only includes one slice and one channel, the alignment can directly be performed using MultiStackReg.
- Correction of lateral drift using MultiHyperStackReg
- Decide which channel and/or slice of the hyperstack should be used as a reference for alignment. To generate a one-channel, one-slice reference stack (henceforth referred to as “reference stack”), click on Image | Duplicate…, tick the Duplicate hyperstack checkbox, type the relevant channel number in Channels (c): (e.g., replace 1–2 with 1) and/or the relevant channel number in Slices (z): (e.g., replace 1–15 with 8), and ensure that Frames: (t) includes all frames (e.g., 1–50) before clicking on OK (Figure 3B).
- Alternatively to step 6.2.1, if a projection of multiple slices is preferred for the one-channel, one-slice reference stack, click on Image | Stacks | Z Project…, select the first and last slices and type of projection, ensure that All time frames is ticked and click on OK. If the time-lapse has multiple channels either delete the irrelevant channels (Image | Stacks | Delete Slice) from the resulting projection or duplicate the chosen reference channel (Image | Duplicate) (Figure 3B).
- Optionally, crop the reference stack to use only one part as the image as a reference by selecting the rectangle tool in the toolbar, tracing a rectangle over the region of interest, and clicking on Image | Crop.
- To start MultiStackReg, click on Plugins | Registration | MultiStackReg. In the pop-up menu Stack 1:, select the name of the one-channel, one-slice stack generated in the previous steps. In the pop-up menu Transformation:, select Translation; tick the Save Transformation File checkbox and ignore all the other fields.
NOTE: Other types of Transformation (see25). - Click on OK. Select a location and name to save the alignment file and click on Save. On the reference stack window, wait for the frames to stop moving automatically, indicating that the registration is over (Figure 3B).
- Check on the reference stack to see whether the alignment is satisfying. If not, repeat steps 6.2.1–6.2.5 with a different reference slice, channel and/or cropping. Close the reference stack.
- Select the hyperstack window and run the MultiHyperStackReg macro. Select the transformation file created in step 6.2.5 and click on Open. Wait for the alignment to be applied to every channel and/or slice of the hyperstack, at which point a new window with the suffix “_aligned” will open (Figure 3C).
- Correction of focus drift using MultiHyperStackReg
- Click on Image | Properties…, copy the value displayed in Pixel Width. To orthogonally reslice the one-channel hyperstack with a one-pixel spacing, click on Image | Stacks | Reslice [/]…, paste the pixel width value into the Output Spacing: numeric field; select Top in the Start At: popup menu, tick Avoid Interpolation and click on OK.
NOTE: In case memory has to be freed on ImageJ, the hyperstack can be closed following the reslice. - Select the new window generated by the reslice (henceforth referred to as “resliced hyperstack”, Figure 4B). If the resliced hyperstack has multiple channels, select a reference channel to perform the alignment and delete the other channels by selecting them in the channel scroll bar; click on Image | Stacks | Delete Slice, select Channel in the Delete Current popup menu and click on OK.
- Generate a single-slice resliced hyperstack (henceforth referred to as “resliced reference stack”), either by duplicating a single slice of the resliced hyperstack (see step 6.2.1) or by projecting several slices of the resliced hyperstack (see step 6.2.2) (Figure 4C).
- Optionally, crop the resliced reference stack to use only one part as the image as a reference (see step 6.2.3).
- Perform the image registration on the resliced reference stack with MutiStackReg (see steps 6.2.4–6.2.6) (Figure 4C).
- Select the resliced hyperstack window and run the MultiHyperStackReg macro. Select the transformation file created in step 6.3.5, click on Open and wait for the alignment to be applied to every channel and/or slice of the hyperstack, at which point a new window with the suffix “_aligned” will open (henceforth referred to as “resliced reference stack”, Figure 4D). Close the original resliced hyperstack.
- To convert the resliced aligned hyperstack to the xy view of the original hyperstack, click on Stacks | Reslice [/]…, select Top in the Start At: popup menu, tick Avoid Interpolation and click on OK. Once a new window with the final focus-aligned hyperstack opens (Figure 4E), close the resliced aligned hyperstack.
- Click on Image | Properties…, copy the value displayed in Pixel Width. To orthogonally reslice the one-channel hyperstack with a one-pixel spacing, click on Image | Stacks | Reslice [/]…, paste the pixel width value into the Output Spacing: numeric field; select Top in the Start At: popup menu, tick Avoid Interpolation and click on OK.
- To automatically correct the lateral drift and focus drift, run the AutoHyperStackReg macro. Select the reference channel, if applicable, and whether to correct the lateral and/or focus drift. Click on OK.
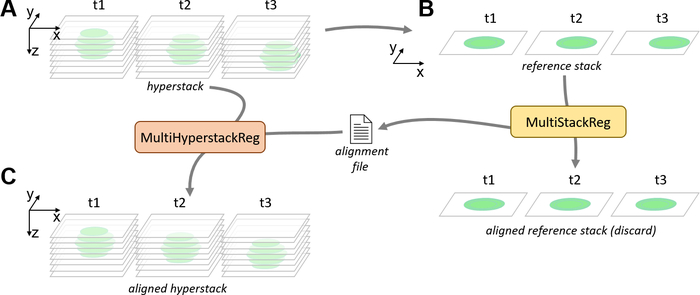
Figure 3: Pipeline for the correction of lateral drift with MultiHyperstackReg. (A) Lateral and focus drifts can be observed on a hyperstack containing several slices and timepoints. (B) A single slice, single channel reference stack is generated from the hyperstack, either by duplication or Z projection. The reference stack is aligned by the MultiStackReg plugin, generating an alignment file. (C) The MultiHyperstackReg macro is used to apply the alignment file to the hyperstack, resulting in an aligned hyperstack for which the lateral drift is corrected. Please click here to view a larger version of this figure.
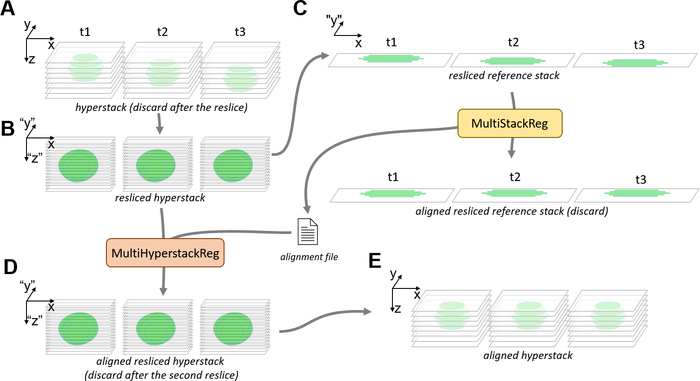
Figure 4: Pipeline for the correction of focus drift with MultiHyperstackReg. (A) Focus drifts can be observed on a hyperstack containing several slices and timepoints. (B) The hyperstack is orthogonally resliced from the top, along each line of pixels along the Y-axis, without interpolation. This results in a resliced hyperstack containing the same information as the hyperstack, with its y and z coordinates switched. The focus drift (in z) of the original hyperstack occurs as a drift in y on the resliced hyperstack. (C) A single slice, single channel resliced reference stack is generated from the resliced hyperstack, either by duplication or Z projection. The resliced reference stack is aligned by the MultiStackReg plugin, generating an alignment file. (D) The MultiHyperstackReg macro is used to apply the alignment file to the resliced hyperstack, resulting in an aligned resliced hyperstack for which the focus drift (in y) is corrected. (E) A second orthogonal reslice restores the original coordinates of the hyperstack, which now no longer presents a focus drift (in z). Please click here to view a larger version of this figure.
7. Cortical signal measurement with rotating linescans
- Download the Rotating Linescans ImageJ macro (Supplemental coding file 3).
- Open a time-lapse in ImageJ. Position the frames scrollbar to the first frame and, in case the time-lapse contains several slices, position the slices scrollbar to the slice on which the signal will be measured.
- Tracking mode
- Select the straight-line tool in the toolbar. Trace a line across the cortical zone to be measured (Figure 5A), making sure that the line is roughly perpendicular to the cortex and that the line is long enough to cross the cortex at every next time point (Figure 5B, first and second panel).
- Double-click the line tool icon in the toolbar to open the Line Width window. Increase the width of the line as much as needed while keeping the intersection between the line and the cortex a straight line (Figure 5B, third panel).
- Start the Rotating Linescans macro. Set the rotation range (Figure 5E) to a low value (about 10°) if the orientation of the cortical zone to be measured does not change much from one timepoint to the next. Set the Measure Around value to 0 pixels to only measure the signal on the line where the maximal signal intensity is detected, or to higher values to also measure the signal around this line.
NOTE: The Measure Around value will only affect the signal measurement after detection of the cortex and will not affect the detection itself. - On the Find Cortex on Channel: popup menu, select the channel on which the detection will be performed. To visualize where the cortex has been detected at each timepoint, keep the Display Position… checkbox ticked (recommended). Keep the Recenter and reorient… checkbox ticked. Click on OK.
- Once the Done, Results Copied to Clipboard status is displayed in the ImageJ window status, scroll through the time-lapse to check whether the detected cortex positions (yellow overlay) and the measured area (cyan overlay) are satisfying. If not, repeat steps 7.3.1–7.3.4 with a different line position, length, or width and different settings in the Rotating Linescans options window.
- Paste the measurements into a spreadsheet software.
NOTE: Columns correspond to channels in the order of their appearance in the time-lapse and lines correspond to frames.
- Non-tracking mode
- Select the straight-line tool in the toolbar. Trace a line (indicated in cyan, Figure 5A) across the cortical zone to be measured, making sure that the line is roughly perpendicular to the cortex and that the line is long enough to cross the cortex at every time point (Figure 5C, first and second panel).
- Double-click on the line tool icon in the toolbar to open the Line Width window. Increase the width of the line as much as needed while keeping the intersection between the line and the cortex a straight line (Figure 5C, third panel).
- Start the Rotating Linescans macro. Set the rotation range (Figure 5E) accordingly to the changes of orientation of the cortical zone to be measured throughout the entire time-lapse. Set the Measure Around value to 0 pixels to only measure the signal on the line where the maximal signal intensity is detected, or to higher values to also measure the signal around this line.
NOTE: The Measure Around value will only affect the signal measurement after detection of the cortex and will not affect the detection itself. - On the Find Cortex on Channel: popup menu, select the channel on which the detection will be performed. To visualize where the cortex has been detected at each timepoint, keep the Display positions… checkbox ticked (recommended). Untick the Recenter and Reorient… checkbox. Click on OK.
- Once Done, Results Copied to Clipboard is displayed in the ImageJ window status, scroll through the time-lapse to check whether the detected cortex positions (yellow overlay) and the measured area (cyan overlay) are satisfying. If not, repeat the previous steps with a different line position, length, or width and different settings in the Rotating Linescans options window.
- Paste the measurements into a spreadsheet software.
NOTE: Columns correspond to channels in the order of their appearance in the time-lapse and lines correspond to frames.
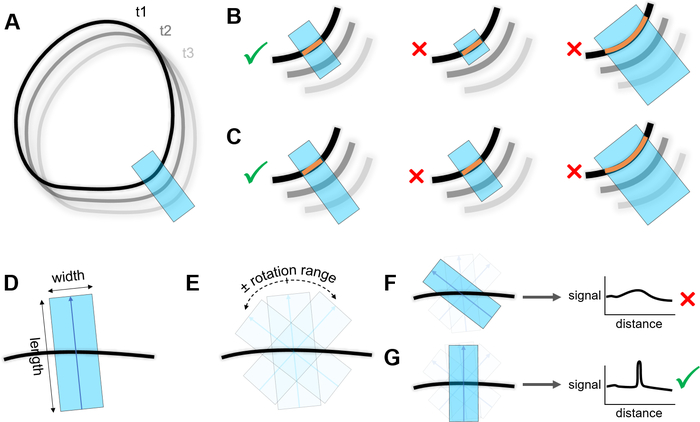
Figure 5: Measurement of cortical signal with rotating linescan. (A) A line (cyan) is traced perpendicularly to the cortex (black) where the signal will be measured. Different shades of gray show the cell position changing over three timepoints (t1, t2, t3). (B) Left: correct positioning of the line in tracking mode. Middle: incorrect positioning of the line in tracking mode as there is no overlap with the cortex at the next timepoint. Right: overly wide line resulting in the intersection (orange line) between the cortex and the line not being straight. (C) Left: correct positioning of the line in non-tracking mode. Middle: incorrect positioning of the line in non-tracking mode as there is no overlap with the cortex at every timepoint. Right: overly wide line resulting in the intersection (orange line) between the cortex and the line not being straight. (D) Length and width of the scanning line. (E) Linescans are performed within a range of orientations around the original orientation shown in (D) defined by the “rotation range” parameter, at an interval defined by the “rotation step” parameter. (F) Non-optimal orientation of the scanning line relative to the cortex, resulting in a low signal measured along the line. (G) The optimal orientation of the line is defined as the one resulting in the highest peak of signal intensity measured along the line. Please click here to view a larger version of this figure.
Immobilization of Drosophila tissues with Fibrin clots and culture medium exchange during live imaging.
After being dissected following the procedure presented in step 2 (Figure 1) and immobilized in Fibrin clots on the same coverslip following the procedure presented in step 3 (Figure 2), larval brains expressing GFP-tagged Bazooka (Baz::GFP), the fly ortholog of the polarity protein Par-3, were imaged for 30 min in time-lapse multi-position confocal imaging. Baz::GFP is combined with apkcas4, an allele that allows acute inhibition of atypical protein kinase C (aPKC) through the addition of the small ATP analog 1-NAPP120. 1-NAPP1 was therefore added to the culture medium following the procedure presented in step 4 and live imaging was resumed for 2.5 h. Control brains carrying a wild-type version of the kinase aPKC did not react to the ATP analog, whereas brains carrying additionally an analog-sensitive mutation of aPKC (apkcas4) displayed a contraction of the neuroepithelium and bright clusters of Baz in neuroblasts and their progeny (Figure 6, Video 1). Neuroblasts keep on dividing throughout the time-lapse, indicating that the tissue remains healthy, and brains show little drift despite the culture medium change, indicating that they are well immobilized. Similarly, after being dissected following the procedure presented in27, ovarioles expressing Baz::GFP were immobilized in Fibrin clots on the same coverslip and imaged for 8 min, after which application of 1-NAPP1 induced the contraction of apkcas4 mutant follicular cells but not of controls (Figure 7, Video 2).
Correction of lateral and focus drift using MultiHyperstackReg.
A hyperstack displaying both lateral and focus drift (Video 3, left panel) was first corrected for lateral drift (step 6.2, Figure 3, Video 3, middle panel), then focus drift (step 6.3, Figure 4, Video 3, right panel) using the MultiStackReg plugin and the MultiHyperstackReg macro, resulting in a substantial reduction of the movements observed in the original hyperstack.
Cortical signal measurement using rotating linescans
A neuroblast expressing Baz::GFP (green) and the transmembrane protein CD4 tagged with an infrared fluorescent protein (CD4::mIFP, red) was imaged during one mitosis. The CD4::mIFP signal was used as a reference to detect the cortex at various parts of the neuroblast with linescans (step 7, Figure 5, Figure 8A–C) and the Baz::GFP signal was measured at the detected positions Figure 8D). During their division, neuroblasts transiently polarized by establishing distinct and opposite cortical domains: the apical pole and the basal pole. Baz defines the apical pole and, consistently, the Baz::GFP signal increased at the apical pole of the neuroblast and decreased at the basal pole during the division (Figure 8C,D). The position of the cortex was satisfyingly tracked throughout the time-lapse (Figure 8C, Video 4). The Drosophila lines and genotypes are referenced in Supplemental Table 1–2.
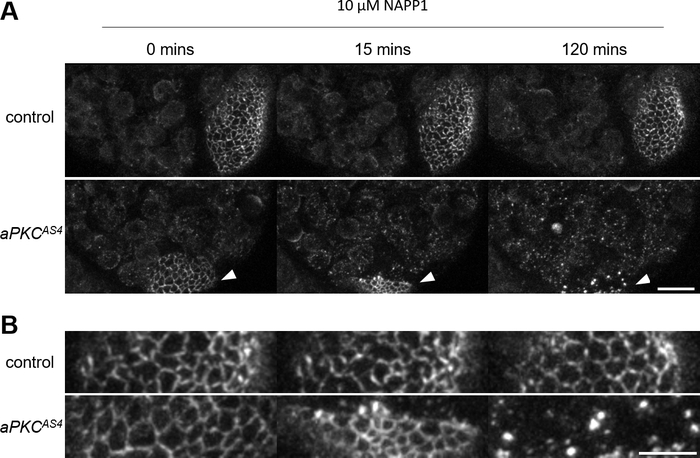
Figure 6: Application of an ATP analog on immobilized larval brains during live imaging. (A) Live imaging of two larval brains expressing Baz::GFP immobilized on the same coverslip. The culture medium is changed to a concentration of 10 µM of the ATP analog 1-NAPP1 at 0 min. Control brains do not visibly react to the ATP analog whereas brains carrying an analog sensitive mutation of aPKC (aPKCAS4) display a contraction of the neuroepithelium (arrowheads) and bright clusters of Baz in neuroblasts and their progeny. Maximal intensity projection of 33 slices for a depth of 23 µM. Scale bar: 20 µM. (B) Magnification of the neuroepithelium. Scale bar: 10 µM. Please click here to view a larger version of this figure.
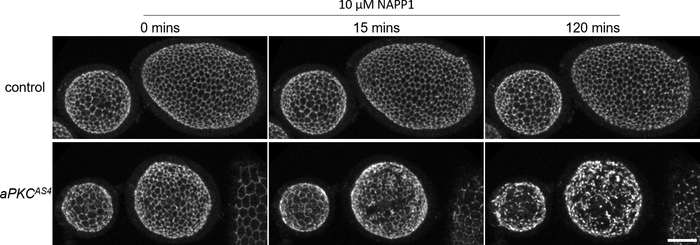
Figure 7: Application of an ATP analog on immobilized egg chambers during live imaging. Live imaging of two ovarioles expressing Baz::GFP immobilized on the same coverslip. The culture medium is changed to a concentration of 10 µM of the ATP analog 1-NAPP1 at 0 min. Control egg chambers do not visibly react to the ATP analog whereas egg chambers carrying an analog sensitive mutation of aPKC (aPKCAS4) display a contraction of the epithelium (arrowheads) eventually leading to the apparent breakdown of adherens junctions. Maximal intensity projection of 33 slices for a depth of 27 µM. Scale bar: 20 µM. Please click here to view a larger version of this figure.
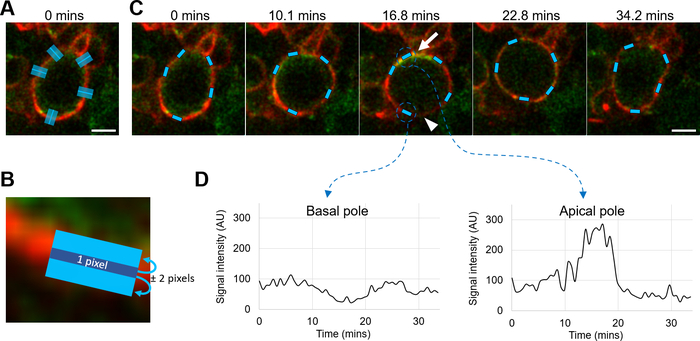
Figure 8: Measurement of the cortical signal using rotating linescans. (A) Live D. melanogaster neuroblast expressing Baz::GFP (green) and CD4::mIFP (red). Cyan lines: initial scanning lines traced perpendicularly to various cortical zones to measure. Scale bar: 5 µM. (B) Identification of the cortex (dark blue line) using cortical linescans and CD4::mIFP (red) as the reference channel. Cyan line: area defined by the “Measure around” parameter (here, 2 pixels), where the signal is measured after cortex detection. Scale bar: 2 µm. (C) Cyan: cortical areas identified during a neuroblast division by the rotating linescans method from the initial scanning lines displayed in (A), using CD4::mIFP (red) as the reference channel. Scale bar: 5 µM. Arrow: apical pole. Arrowhead: basal pole. (D) Measurement of the Baz::GFP signal intensity over time in the two corresponding zones identified by rotating linescans displayed in (C). During mitosis, Baz::GFP gets transiently enriched at the apical pole of the neuroblast and is depleted from the opposite basal pole. Please click here to view a larger version of this figure.
Video 1: Application of media exchange on immobilized larval brains during live imaging to add an inhibitor, presented in Figure 6, scale bar: 20 µm. Please click here to download this video.
Video 2: Application of media exchange on immobilized egg chambers during live imaging to add an inhibitor, presented in Figure 7, scale bar: 20 µm. Please click here to download this video.
Video 3: Correction of lateral and focus drift using MultiHyperStackReg. Live imaging of a neuroblast expressing the membrane probe PH::GFP. Xy: single focal plane. Xz: orthogonal reslice. Left: before correction of the drift. Middle: after correction of the lateral drift. Right: after correction the lateral and focus drift, scale bar: 10 µm. Please click here to download this video.
Video 4: Detection of the cortex using rotating linescans, presented in Figure 8, scale bar: 5 µm. Please click here to download this video.
Supplemental Table 1: Genotypes of imaged Drosophila tissues. Please click here to download this table.
Supplemental Table 2: Origin of transgenes used. Please click here to download this table.
Supplemental Coding File 1: Origin of transgenes used. Please click here to download this file.
Supplemental Coding File 2: Origin of transgenes used. Please click here to download this file.
Supplemental Coding File 3: Origin of transgenes used. Please click here to download this file.
