A schematic flow chart of this procedure (Figure 1) visually summarizes the steps that were taken to successfully fractionate U9375 cells grown in suspension. Fractions collected from the top of the isopycnic density gradient in equal volumes (1 mL) show the purification of the mitochondrial and membrane fractions (Figure 2). Utilizing an antibody against VDAC, a protein localized to the outer mitochondrial membrane6, shows that the mitochondrial fraction migrated to the 25% and 30% iodixanol (v/v) fractions (Figure 2A). Using an antibody against the Na,K+-ATPase α1 subunit, part of an integral membrane heterodimer found primarily in the plasma membrane7, shows the separation of membrane contamination from the pure mitochondrial fraction (Figure 2A). The pure membrane fraction migrated to the least dense fractions, 10% and 15% iodixanol (v/v) (Figure 2B). Mitochondrial contamination of the membrane fraction was separated by the gradient.
A western blot8 performed with the additional localization markers (referenced in step 7.4) shows the purity of the cytosolic and nuclear fractions, while additionally verifying that the mitochondrial and membrane samples are free from contamination by proteins from other parts of the cell (Figure 3). Using an antibody against GAPDH, normally localized to the cytoplasm of the cell9, shows that this protein is only found in the cytosolic fraction (Figure 3A, Lane 1, first panel), and that no contamination is observed in the extracted nuclear proteins, the density-purified mitochondria, or membrane fractions (Figure 3A; Lanes 2, 3, and 4; first panel). Probing for histone H3, a protein found in the nucleus and involved in chromatin structure10, shows a successful nuclear extraction (Figure 3A, Lane 2, second panel), with some minimal detection in the cytoplasmic fraction and no cross contamination in the mitochondrial or membrane fractions.
Probing for VDAC in all fractions shows the presence of this protein in the pure mitochondrial fraction (Figure 3A, Lane 3, third panel), and that no cross contamination exists in the other fractions (Figure 3A; Lanes 1, 2, and 4; third panel). Probing for the Na/K-ATPase α1 subunit similarly shows that this protein is located only in the pure membrane fraction (Figure 3A, Lane 4, fourth panel). These fractions were analyzed by densitometry to confirm reproducibility and statistical significance (Figure 3B–E). In contrast to the results from the successful fractionation (Figure 3), improper execution of this method (or failure to adhere to all recommended steps) can result in cross-contamination of cellular components (Figure 4). A high concentration of histone H3 in the cytosolic fraction (Figure 4, Lane 1, second panel) can result from a failure to properly clarify the cytosolic fraction (referenced in step 2.6). This can occur if not enough clarification centrifuge spins are performed, or if the cytosolic fraction is not clarified quickly. If the cytosolic fraction is not clarified quickly enough, it may result in lysis of cell fragments, leading to contamination of the cytosolic fraction.
Failure to perform the isopycnic density purification step will result in contamination of the membrane fraction (Figure 4, Lane 4, all panels), depending on how heterogenous the sample is prior to density purification. Proper adherence to all steps of the protocol is critical to obtaining the desired separation of the subcellular fractions. When quantified using a Bradford assay, protein yield for each fraction can be determined. Expected protein yield per fraction is reported in Table 1. It is useful to perform a protein quantification assay prior to performing western blots for several reasons. First, it confirms that fractions indeed contain protein; second, it allows loading of SDS-PAGE gels based on protein quantity; and finally, assuming protein yields are similar to those expected (Table 1), it confirms proper execution of the procedure.
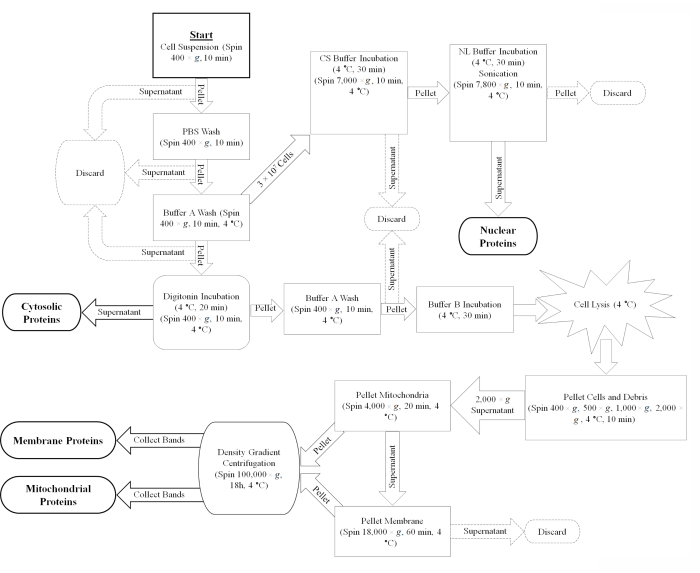
Figure 1: Diagram of the cell fractionation procedure. An overview of the cell fractionation protocol represented as a flow chart. Abbreviations: PBS = phosphate-buffered saline; CS = cell solubilization; NL = nuclear lysis. Please click here to view a larger version of this figure.
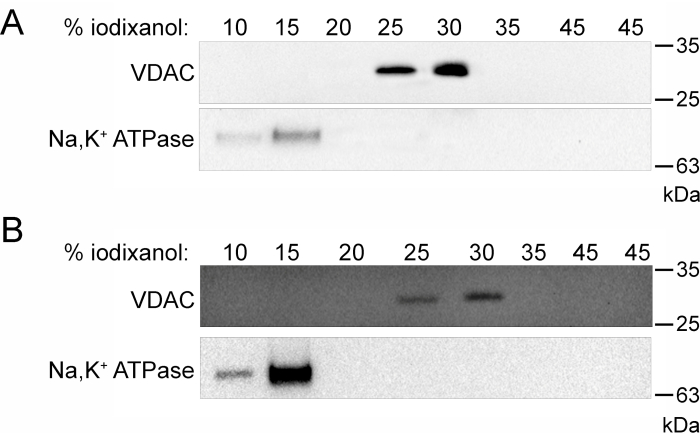
Figure 2: Isopycnic density purification of crude mitochondrial and membrane fractions. (A) Representative western blot of all fractions collected after density gradient purification of the crude mitochondrial fraction. (B) Representative western blot of all fractions collected after density gradient purification of the crude membrane fraction. Both density gradient purifications show migration of the mitochondrial marker, VDAC, for the 25% and 30% iodixanol (v/v) fractions and migration of the membrane marker, Na,K+ ATPase, for the 10% and 15% iodixanol (v/v) fractions. Abbreviation: VDAC = voltage-dependent anion channel. Please click here to view a larger version of this figure.
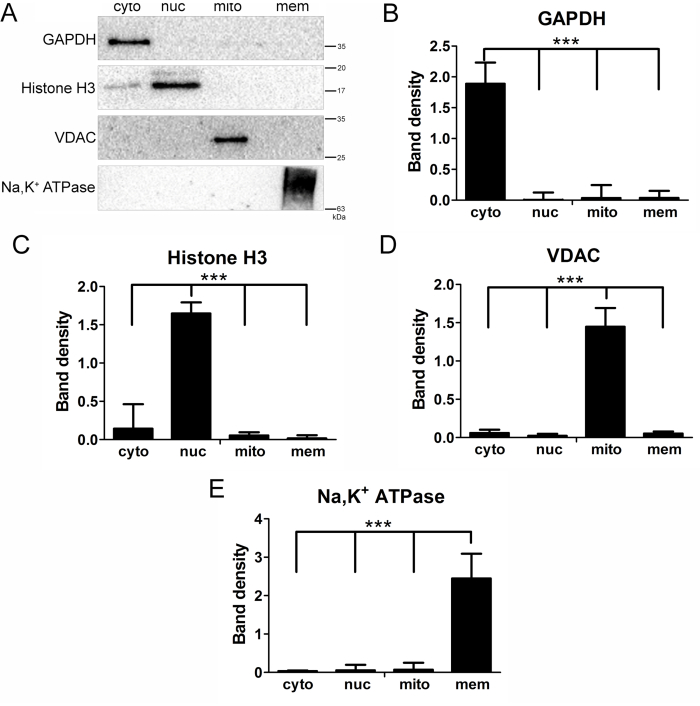
Figure 3: Successful isolation of U937 cytosolic, nuclear, mitochondrial, and membrane fractions. (A) Representative western blots of cell fractions isolated from a U937 cell culture with this technique and probed for markers of cytoplasm (GAPDH, first panel), nucleus (Histone H3, second panel), mitochondria (VDAC, third panel), and membrane (Na,K+ ATPase α1, fourth panel). (B–E) Densitometry of western blots of cell fractions isolated from U937 cell cultures with this technique. Results are from 3 independent experiments. Error bars represent standard deviation. One-way ANOVA. ***p<0.001. Abbreviations: GAPDH = glyceraldehyde-3-phosphate dehydrogenase; VDAC = voltage-dependent anion channel; cyto = cytosolic; nuc = nuclear; mito = mitochondrial; mem = membrane. Please click here to view a larger version of this figure.
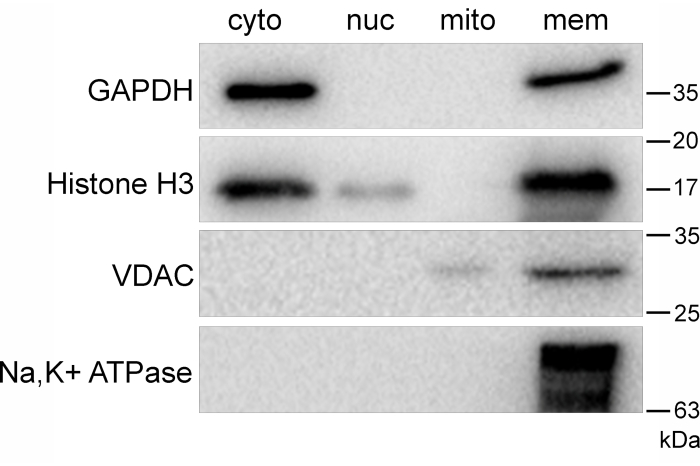
Figure 4: Incomplete fractionation of U937 cell components. Representative western blots of cell fractions isolated from a U937 cell culture showing contamination of the cytosolic fraction with histone H3 (Lane 1, second panel) due to improper clarification of this fraction and a crude membrane fraction that was not subjected to isopycnic density gradient purification (Lane 4, all panels). Abbreviations: cyto = cytosolic; nuc = nuclear; mito = mitochondrial; mem = membrane. Please click here to view a larger version of this figure.
| Fraction | Yield (µg/mL) | Standard Deviation | Approximate Volume |
| Cytoplasm | 1500 | 146 | 5 mL |
| Nucleus | 1200 | 172 | 500 µL |
| Mitochondria | 400 | 66 | 500 µL |
| Membrane | 200 | 23 | 500 µL |
Table 1: Protein yield for each subcellular fraction.
