Microglia are brain tissue-resident macrophages, and their functions include immune surveillance, coordinating inflammatory responses to injury/infection, synaptic remodeling, and phagocytosis of dead cells, myelin, protein aggregates, and pathogens. Phagocytosis is the process by which microglia recognize cargo with surface receptors and reorganize their cytoskeleton to engulf the object into a phagosome, which then fuses with lysosomes for degradation of the cargo. Healthy microglia phagocytose apoptotic brain cells to remove them before they become necrotic1. The phagocytosis of apoptotic cells is also known as efferocytosis, and requires the display of a phosphatidylserine "eat-me" signal by the dying cell2. Numerous microglia receptors directly bind to phosphatidylserine, including TIM-4, BAI1, Stabilin-2, and TREM2. Microglial TAM receptors (e.g., MERTK) and integrins indirectly bind to phosphatidylserine, using accessory proteins GAS6 or MFG-E8, respectively. Other "eat-me" signals may be necessary for recognition of dying cells, these include changes to glycosylation or charge of surface proteins; expression of intracellular proteins ICAM3, calreticulin, annexin-I at the cell surface; oxidized LDL; or coating of the apoptotic cell by microglia-produced complement C1q1,2.
Neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis have been associated with impairment to microglia function, including an accumulation of brain waste products such as dead cells, myelin fragments, and protein aggregates, and exaggerated inflammatory responses to these stimuli3. Phagocytosis may be impaired in neurodegenerative diseases and contribute to the pathology, due to a combination of aging, inflammation, or specific genetic risk variants4,5. On the other hand, there is also evidence from animal models of neurodegenerative diseases that microglia may inappropriately phagocytose viable neurons or synapses6,7,8. The mechanism is likely to be instigated by phosphatidylserine display of damaged neurites, which is directly sensed by microglial phagocytosis receptors TREM2 or GPR56, or indirectly sensed by soluble complement C1q coating the phosphatidylserine-enriched membrane, leading to CR3-mediated phagocytosis9,10,11.
In vitro assays of phagocytosis function, e.g., to assess the phenotypic impact of a genetic risk variant in microglia, are frequently performed using non-physiological cargoes such as latex beads4. Fluorescently labeled bacteria and zymosan are also used, which are physiological but not relevant to neurodegenerative diseases. Non-physiological phagocytic cargoes can be used to detect defects in the basic machinery of phagocytic engulfment but fail to accurately model the first "recognition" step in phagocytosis of apoptotic neurons. The size, shape, stiffness, and type of cargo also dictate the intracellular signaling pathways that are activated, leading to different outcomes of microglia activation state. For example, E.coli bacteria are small and stiff, unlike human cells, and the lipopolysaccharides on their surface are recognized by Toll-like receptor 4 (TLR4) that activates phagocytosis and pro-inflammatory signaling pathways2,12.
In the context of neurodegenerative disease studies, a more relevant phagocytic cargo would have phosphatidylserine display on mammalian plasma membranes, and would ideally be human and neuronal, to include signals that microglia are likely to encounter. For this phagocytosis protocol, the human neuroblastoma cell line SH-SY5Y was chosen as a neuron model that is easy to culture. Permanent surface phosphatidylserine display was artificially induced by paraformaldehyde, which has previously been shown to cause phosphatidylserine display of platelets13. For the microglia cell model human iPSC-macrophages were used, which mimic the ontogeny and transcriptional profile of human microglia, and are phagocytically competent14,15,16,17. iPSC-macrophages are not the most authentic microglia model available, e.g., they do not mimic microglia morphology; however, one can substitute it for a more authentic monoculture iPSC model of microglia if desired, such as Haenseler et al.15. Human iPSC models are preferable to primary rodent microglia for studying neurodegeneration, due to concerns about the limited overlap of the microglia transcriptional modules observed in human versus mouse neurodegenerative disease tissues18. The dead SH-SY5Ys are stained with an acid-sensitive dye that fluoresces weakly at neutral pH and more strongly inside the phagolysosomes of iPSC-macrophages after phagocytosis. Using an acid-sensitive dye improves the accuracy of detecting phagocytic events, with versatility for different readouts of live and fixed macrophages19. This protocol outlines both live-cell time-lapse imaging of phagocytosis, and a fixed high-content imaging assay for phagocytosis, with the same cell preparation steps prior to readout (Figure 1).
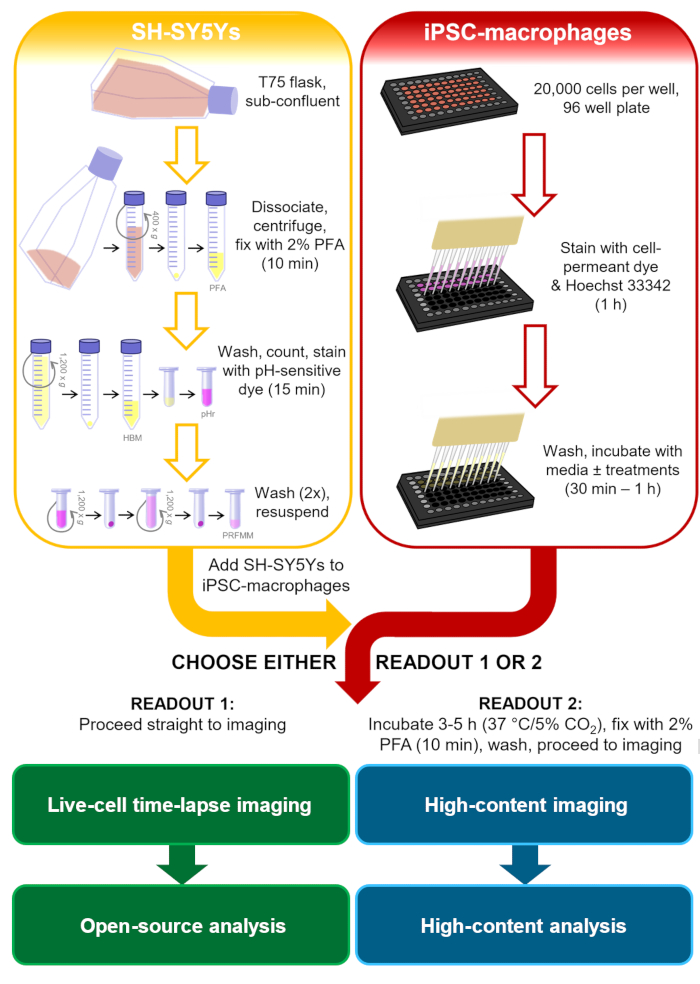
Figure 1: Schematic diagram of methodology. Outline of the phagocytosis assay, where preparation of the SH-SY5Ys and staining of the iPSC-macrophages is performed in parallel, and then the SH-SY5Ys are pipetted onto the iPSC-macrophages. Either live-cell time-lapse imaging is performed immediately, or the cells are incubated at 37 °C/5% CO2 for the required duration and fixed before performing high-content microscopy. PFA: paraformaldehyde, HBM: phenol red-free HEPES-buffered media, pHr: pH-sensitive red fluorescent dye STP Ester solution, PRFMM: phenol red-free macrophage media. Please click here to view a larger version of this figure.
The protocol follows the guidelines for the use of human iPS cell lines derived at the University of Oxford, Oxford Parkinson's Disease Centre (Ethics Committee: National Health Service, Health Research Authority, NRES Committee South Central, Berkshire, UK (REC 10/H0505/71)). Human iPSCs are to be handled within a Class II safety cabinet to protect the worker from possible adventitious agents. Local, national, and EU health and safety regulations must be adhered to. Cell culture media compositions are detailed in Table 1, and all materials are listed in the supplementary Table of Materials.
1. Cell culture prior to experiment
- Culture iPSCs in iPSC media (Table 1) in 6-well plates pre-coated with a hESC-qualified basement membrane matrix, sub-confluent and at a low passage number.
- Differentiate human iPSCs to iPSC-macrophage precursors: seed four million iPSC into a microwell low-adherence 24-well plate with 2 mL of embryoid body media (Table 1) to encourage embryoid body formation and perform 75% media changes daily for 5-6 days. Transfer embryoid bodies into T175 flasks, approximately 150 embryoid bodies per flask, containing 20 mL of factory media (Table 1). Feed weekly by addition of 10-20 mL of factory media.
NOTE: iPSC-macrophage precursors emerge into the supernatant after approximately 2-3 weeks and are produced continuously for several months. For this experiment, it is preferable to use cells from approximately 6 weeks after the differentiation factories are set up. Earlier harvested iPSC-macrophages can retain some proliferative capacity and are less adherent, preventing even seeding at a low cell density. An upper age limit for phagocytosis capability has not been determined. - Differentiate iPSC-macrophage precursors to iPSC-macrophages: harvest precursors by removing the required volume of supernatant; pass it through a 40 µm cell strainer to remove clumps; centrifuge at 400 x g for 5 min to pellet cells and resuspend in macrophage media (Table 1). Seed iPSC-macrophages at 20,000-30,000 cells per well in a 96-well tissue culture (TC)-treated microplate with black well walls and an optically clear bottom, in 100 µL of macrophage media per well. Avoid the edge wells and fill these with PBS; this is important to reduce the effect of evaporation on the assay. Differentiate for 6-10 days by incubation at 37 °C/5% CO2.
NOTE: For this assay, the iPSC line BIONi010-C (ECACC ID: 66540023) was used; however, another iPSC line can be substituted. - Maintain SH-SY5Ys to sub-confluency in T75 flasks with 20 mL of SH-SY5Y media (Table 1), passaging every 3-4 days.
| Name | Base media | Additive, final concentration |
| iPSC media | mTeSR1 | – |
| Embryoid Body media | mTeSR1 | BMP4, 50 ng/mL |
| VEGF, 50 ng/mL | ||
| SCF, 20 ng/mL | ||
| Factory media | XVIVO15 | GlutaMAX, 2 mM |
| Penicillin, 100 units/mL | ||
| Streptomycin, 100 µg/mL | ||
| 2-Mercaptoethanol, 50 µM | ||
| IL-3, 25 ng/mL | ||
| M-CSF, 100 ng/mL | ||
| Macrophage media | XVIVO15 | GlutaMAX, 2 mM |
| Penicillin, 100 units/mL | ||
| Streptomycin, 100 µg/mL | ||
| M-CSF, 100 ng/mL | ||
| SH-SY5Y media | DMEM/F12 | Fetal bovine serum, 10% |
| Penicillin, 100 units/mL | ||
| Streptomycin, 100 µg/mL |
Table 1: Media recipes.
Constituents of cell culture media used in the protocol. Further details of the media components can be found in the Table of Materials.
2. Preparation of dead SH-SY5Ys
- In a class II biological safety cabinet, dissociate SH-SY5Ys, by addition of 4 mL of a cell dissociation buffer containing recombinant trypsin-like enzymes and 1.1 mM EDTA (see Table of Materials), which should be removed immediately so that less than 1 mL remains as a thin film coating the cells. Incubate for 2-3 min at 37 °C/ 5% CO2.
- Add 10 mL of HBSS to the T75 flask to rinse, and pipette the SH-SY5Ys into a 15 mL conical centrifuge tube. Centrifuge at 400 x g for 5 min. Aspirate the supernatant and re-suspend the cells in 2 mL of phenol red-free HEPES-buffered media (see Table of Materials). Ensure to resuspend the pellet carefully, pipetting with a 100-1,000 µL pipette to break up clumps before fixation.
- Fix cells by adding 2 mL of 4% paraformaldehyde (final concentration 2%) to the tube. Incubate for 10 min at room temperature with occasional gentle agitation of the tube.
- Add 10 mL of HBSS to the tube. Centrifuge at 1,200 x g for 7 min and re-suspend in 2 mL of phenol red-free HEPES-buffered media.
NOTE: After step 2.4, the fixed-SH-SY5Y preparation can be quality-controlled by staining with annexin V-FITC to show accessible phosphatidylserine and propidium iodide to measure cell permeability with a flow cytometry readout. Compare the fixed preparation to live SH-SY5Ys obtained from step 2.2. See section 7 and Supplementary Figure S1. Storage of the fixed SH-SY5Ys after step 2.4 is not recommended as this has not been assessed.
3. Labeling of dead SH-SY5Ys with pH-sensitive red fluorescent dye
- After step 2.4, count the cells, and remove the total number of cells required into a 2 mL low protein-binding tube. For each 1 million SH-SY5Ys, make up the total volume in the 2 mL tube to 300-500 µL with phenol red-free HEPES-buffered media. Warm the tube briefly in a 37 °C water bath.
- Reconstitute the pH-sensitive red fluorescent dye STP ester (see Table of Materials) and add 12.5 µg of dye per million SH-SY5Y to the warm 2 mL tube of cells. Mix gently by pipetting. Incubate the tube at room temperature for 30 min, protected from light.
NOTE: The STP ester species of the pH-sensitive dye reacts with primary amines and therefore the labeling buffer must not contain free amines. Due to potentially limited solubility in aqueous buffers, add the DMSO-dissolved dye only to warm aqueous buffer, mix immediately, and examine for signs of precipitate (dark particles under a light microscope). - Add 1 mL of HBSS and centrifuge at 1200 x g for 7 min at 4 °C. Discard the supernatant and wash with 2 mL HBSS. Repeat the centrifugation.
- Discard the supernatant and re-suspend the cells in phenol red-free macrophage media (see Table of Materials), to a concentration of 200,000-1.2 million cells/mL so that 50 µL is 10,000-60,000 cells (i.e., 0.5x-3x more SH-SY5Ys than iPSC-macrophages).
NOTE: Phenol red in media increases background fluorescence and therefore a phenol red-free media should be used if live-cell imaging is to be performed. Storage of the stained SH-SY5Ys for more than a few hours is not recommended as this has not been assessed. Keep stained SH-SY5Ys on ice and protect from light.
4. Staining of iPSC-macrophages
- In a biological safety cabinet, prepare a solution in macrophage media of a deep red-fluorescent, cell-permeant, succinimidyl ester-reactive dye (see Table of Materials). Add Hoechst 33342 (see Table of Materials). Warm the working solution to 37 °C in a water bath.
- Aspirate the iPSC-macrophage medium gently by pipetting the cell supernatant with a multichannel pipette into a sterile reservoir. Add 70 µL/well of the dye solution prepared in step 4.1 to iPSC-macrophages, using a multichannel pipette. Incubate for 1 h at 37 °C/5% CO2.
- Prepare experimental treatments in phenol red-free macrophage media. Include 10 µM cytochalasin D as a negative control treatment. Post-incubation aspirate the iPSC-macrophage medium very gently with a multichannel pipette, and add 100 µL/well of Hank's buffered saline solution (HBSS) to wash. Immediately remove HBSS by gentle pipetting, then add 100 µL of media ± compounds. Incubate for 10 min-1 h at 37 °C/ 5% CO2.
NOTE: Cytochalasin D is a potent actin inhibitor and blocks phagocytosis. For any experimental treatments that require longer incubation, e.g., 24-72 h, perform the experimental treatment before step 4.1 using 100 µL/well of treatment in full macrophage media. Follow steps 4.1-4.3 as per the protocol so that cell staining is performed and subsequently the treatment is reapplied in phenol red-free macrophage media for the remainder of the phagocytosis assay.
5. Imaging phagocytosis
Below are two different phagocytosis readout methods, choose sub-section 5.1 or 5.2.
- Live-cell time-lapse imaging
- Prior to phagocytosis, turn on the live-cell time-lapse imaging microscope (see Table of Materials), computer, environment chamber, and CO2 gas. Open image capture software. Check that the DAPI, RFP, and CY5 light cubes are installed in the microscope. Click on Time Lapse | Incubate | Enable Environment Chamber and select warming to 37 °C with CO2 gas, also ensure that humidity is de-selected. Allow 30 min for the microscope to warm to 37 °C.
- During the compound incubation at step 4.3, load the iPSC-macrophage plate into the microscope.
- Click on Image | Capture | Vessel Expert. Select Well Plate and choose a 96-well plate type.
- In the Image tab, turn on the phase channel and adjust coarse and fine focusing using the vertical sliders, so that the cells are in focus. Adjust lighting levels with the horizontal slider. Click on the DAPI, RFP, and CY5 channels and adjust lighting levels for each channel.
- In the System tab, click on Calibrate Vessel Alignment and follow the screen instructions.
- Click on Time Lapse | Routines | Create New Routine. On the first screen of the Time Lapse Wizard, name the routine. Click on Next. On the second screen, select the 20x objective, select Monochrome capture, and select the DAPI, RFP, CY5, and Phase channels. Do not select the following options: Auto Find Sample, Auto fine focus, Z-Stack, Auto lighting. Click on Next.
- On the next screen set up a beacon in the center of each well, which will allow the microscope to return to the same co-ordinates with the same lighting settings for every time-point. The focusing and lighting settings for each beacon are independent. To set a beacon: drag the blue circle to the location on the plate map, use the coarse and fine focus vertical slider, and when satisfied click on Add Beacon. Beacon settings can be updated later using the Update Selected button.
- When ready to start the phagocytosis assay, remove the assay plate and place it in a biological safety cabinet. Use a multichannel pipette to add 50 µL of SH-SY5Ys per well, adding to the side of each well at the edge of the liquid.
- Load the plate into the microscope and wait approximately 30 min for the thermal shift to equilibrate.
NOTE: During the first 30 min that the plate is in the microscope, the changing temperature of the assay plate will cause the focus to shift. If the plate is not allowed to equilibrate, the captured images will shift out of focus during the time-lapse. - Click on each beacon and update the focus setting. Click on Next. On the next screen of the Time Lapse Wizard, select the file format TIFF, enable the option to Save Individual Channels, and enable the option to Create Video for Each Beacon, and allow the options beneath Include the following information as a watermark. Click on Next.
- Set the Number of Scenes to 1. Click on Next. Set the duration and intervals of the time-lapse, e.g., 3 h and imaging every 5 min. Do not select Capture one frame only. Click on Next.
- Enable the environment chamber, with a temperature of 37 °C and CO2 (humidity is optional for short experiments). Click on Next twice. Choose a path to save the data. Click on Next. Click on Start to start the time-lapse.
- Fixed-cell high-content imaging
- Use a multichannel pipette to add 50 µL of the labeled SH-SY5Ys per well, adding to the side of each well at the edge of the liquid. Incubate at 37 °C/ 5% CO2 for 3-5 h.
- After the phagocytosis incubation, gently aspirate cell supernatants by pipetting with a multichannel pipette, and discard. Wash once with 100 µL PBS.
- Fix the plate by addition of 100 µL of 2% paraformaldehyde, incubate for 15 min at room temperature.
- Aspirate wells and add 100 µL of PBS. Cover with plate sealer and foil; store at 4 °C until required.
NOTE: The assay plate can be stored like this for at least a week without significant signal degradation; longer storage has not been tested. - Turn on the high-content imaging microscope (see Table of Materials) and open the image capture software. Load the assay plate into the microscope by clicking on the Load icon at the top of the screen.
- Select the Setup tab. In drop-down menus of the top left box: select the appropriate plate type, select the autofocus option Two Peak (Default), select the objective 40x Water, NA1.1, select Confocal mode, and select binning of 1.
- Flush the 40x water objective before use, via the Settings menu.
- In the Channel Selection box, use the + icon to add the channels DAPI, Alexa 647, and Alexa 568. Set these to measure at a single plane of 1 µm. Optimize Time and Power settings for the staining efficiency of the assay plate.
NOTE: As a guideline, set DAPI at 200 ms exposure and 100% power, Alexa 647 at 1500 ms exposure and 100% power, and Alexa 568 at 100 ms exposure and 40% power. - Ensure the channels are not measured simultaneously by clicking on Channel Sequence to separate out the channels.
- Under Navigation | Define Layout, select the wells of measurement and select 9-12 fields per well.
- During set-up, click on a representative field on the plate map, and check each measurement channel in turn, to ensure the staining is present and that the images are focused, by adjusting the channel offset.
- For data to be uploaded to a server for remote analysis, click on the Online Jobs box and the relevant screen name; this will enable automatic uploading of the data to a server after imaging.
- Save the assay protocol by clicking on the Save button.
- Click on the Run Experiment tab at the top and name the experiment plate, then click on Start.
6. Data analysis
Below are two different data analysis methods, choose sub-section 6.1 if sub-section 5.1 was followed, or choose sub-section 6.2 if sub-section 5.2 was followed.
- Analysis of phagocytosis images obtained by live-cell time-lapse microscope
- Download and install the recommended open-source software (see Table of Materials). Open the software.
- In the Input modules box, select Images.
- From Windows Explorer, open the folder of data, containing sub-folders named Beacon-1, Beacon-2, etc. Select and drag all of the Beacon folders into the File list box.
- In the Input modules box, select Metadata. For Extract Metadata?, select Yes. In the drop-down menu next to Metadata Extraction Method, choose Extract From File/Folder Names. For the Metadata Source, choose Folder Name. Click on the magnifying glass to the right of regular expression, and type ".*[.*](?P<Beacon>.*)$" into the Regex textbox (excluding the quote marks). Click on Submit. For Extract Metadata From, choose All Images. Click on Update at the bottom of the screen. The images will now be grouped by beacon.
- In the Input Modules box, select NamesAndTypes. The following process will allow images for each timepoint to be assigned to the correct fluorescence channel. Assign a name to Images Matching Rules (drop-down menu). Select the rule criteria match All (drop-down menu) of the following rules. File (drop-down menu), Does (drop-down menu), Contain (drop-down menu), DAPI (text box). Name to assign to these images DAPI (text box). Select the image type Greyscale Image (drop-down menu). Set intensity range from Image Metadata (drop-down menu).
- At the bottom of the screen, click on Add Another Image, and repeat step 6.1.5. Replace DAPI with RFP, so that the RFP images are grouped.
- Repeat step 6.1.6 for the CY5 channel images.
- Click on Update at the bottom of the screen, the image files will now be listed in three columns labeled DAPI, RFP, and CY5.
- In the Input Modules box, select Groups. For Do you want to group your images?, select Yes. In the drop-down menu for Metadata Category, choose Beacon.
- In the Analysis Modules box, right-click on the white space to call up a list of all modules.
- Click on Add | Image Processing | EnhanceOrSuppressFeatures. Choose DAPI from the first drop-down box as the input image. Name the output image as "DAPIspeckles". Select the operation type Enhance and feature type Speckles, with a feature size of 20 pixels. Choose the speed and accuracy option Fast/Hexagonal.
- Create a new module. Add | Object processing | IdentifyPrimaryObjects. Choose DAPIspeckles from the first drop-down box as the input image. Name the primary objects "Nuclei". Input the typical diameter of objects as 10 to 35 pixel units; this parameter can be optimized. Choose the threshold strategy Global, the thresholding method RidlerCalvard, the smoothing method Automatic, and give the threshold correction factor as 12 with lower and upper bounds 0-1. Change the method to distinguish clumped objects to Shape but leave other parameters at their default settings.
NOTE: The iPSC-macrophage nuclei have been roughly segmented in step 6.1.12, following an image processing step that reduces diameter and increases contrast of the nuclei. It is important that only the brightest nuclei are selected since the SH-SY5Ys will show up as fainter nuclei and be mistaken as iPSC-macrophages otherwise. To adjust the proportion of nuclei that are selected, increase or decrease the threshold correction factor. During the testing phase, compare the resulting nuclei selection to a phase image of the beacon, where it is easy to distinguish between iPSC-macrophage and SH-SY5Y using cell morphology. - Create a new module. Add | Image processing | CorrectIlluminationCalculate. Choose CY5 from the first drop-down box as the input image. Name the output image "IllumCY5". For Select How the Illumination, choose Background from the drop-down menu. Leave the other parameters at their default settings.
- Create a new module. Click on Add | Image processing | CorrectIlluminationApply. Choose CY5 from the first drop-down box as the input image. Name the output image "CorrCY5". For Select the Illumination, choose IllumCY5 from the drop-down menu. For Select How the Illumination, choose Divide from the drop-down menu.
NOTE: The purpose of steps 6.1.13-6.1.14 is to correct variation in background lighting of the CY5 images, which would otherwise interfere with correct cell segmentation. - Create a new module. Click on Add | Object processing | IdentifySecondaryObjects. Choose CorrCY5 from the first drop-down box as the input image. Choose Nuclei as the input objects. Name the secondary objects "Mac". Choose the method of identification as Distance – B. Select threshold strategy Global, thresholding method RidlerCalvard, the smoothing method No smoothing, and give the threshold correction factor as 1 with lower and upper bounds 0-1. Leave other parameters at their default settings.
NOTE: This cell segmentation step may require optimization, by adjusting the threshold correction factor to grow or shrink cell boundaries. Segmentation efficiency can also be improved by increasing the strength of the iPSC-macrophage staining, or the illumination of the CY5 light cube during imaging. - Create a new module. Click on Add | Image processing | EnhanceOrSuppressFeatures. Choose RFP from the first drop-down box as the input image. Name the output image as "FilteredRFP". Select the operation type Enhance and feature type Speckles, with a feature size of 15 pixels. The feature size can be optimized. Choose the speed and accuracy option Fast/Hexagonal.
- Create a new module. Click on Add | Object processing | IdentifyPrimaryObjects. Choose FilteredRFP from the first drop-down box as the input image. Name primary objects "pHr". Input the typical diameter of objects as 5-20 pixel units. Select the threshold strategy Manual, and type a threshold manually, e.g., 0.005. Change the method to distinguish clumped objects to Shape but leave the other parameters at their default settings.
NOTE: The SH-SY5Ys have been segmented in step 6.1.17, following an image processing step that reduces diameter and increases contrast of the puncta. It is critical to perform manual thresholding, since the intensity of the pH-sensitive dye increases over time in phagocytosed particles, and other thresholding strategies will artificially inflate the number of pH-sensitive dye puncta in early time points. Manual thresholding must be adjusted for each subsequent experimental repeat, using the test mode. - Create a new module. Click on Add | Object processing | RelateObjects. Select the input child objects pHr from the drop-down menu. Select the input parent objects Mac from the drop-down menu. For Calculate Per-Parent Means for All Child Measurements?, select Yes. Do not calculate child-parent distances (None).
NOTE: Step 6.1.18 relates the pH-sensitive dye signal to the iPSC-macrophages, allowing measurement of the average number of phagocytosed objects per iPSC-macrophage. - Create a new module. Click on Add | File processing | ExportToSpreadsheet. Select the column delimiter as Tab and add a prefix for file names to indicate the beacon number. Choose specific measurements for export, as indicated below (steps 6.1.19.1 – 6.1.19.4); leaving other parameters at their default settings.
- Image | Count | Select pHr and Mac
- Image | FileName
- Image | Group
- Mac | Children | pHr
- In the Output box, click on View Output Settings. Create a new folder on the desktop for this experiment and set this as the default output folder.
- Save the pipeline File | SaveProject As….
- Test and optimize the pipeline on a representative image by clicking on Start Test Mode in the bottom-left corner. The program automatically selects the first image for testing and each step of the pipeline can be viewed by clicking on the eye symbols, which makes the output visible, and then clicking on Run. To change the beacon used for testing, at the top menu bar click on Test | Choose Image Group. To change the image (timepoint) within a beacon, at the top menu bar click on Test | Choose Image Set. Parameters that should be optimized are stated in the previous steps.
- When satisfied with the pipeline, click on Exit Test Mode and click on the open eye symbols to close them. Save the pipeline. Click on Analyze Images to start the full image analysis.
- The text files that are generated can be opened as a spreadsheet with appropriate spreadsheet software, and the file labeled "Image" will contain a row for each image timepoint, with the columns representing parameters.
NOTE: Count_Mac and Count_pHr represent the number of iPSC-macrophages and the number of identified pH-sensitive objects in an image. Do not use Count_pHr data, as the count includes dimly fluorescent SH-SY5Ys that have not been phagocytosed. The column Mean_Mac_Children_pHr_Count takes the average number of phagocytosed pHr objects per Mac (step 6.1.18 RelateObjects) for an individual image, i.e., individual timepoint of a beacon. - Arrange the data so that each beacon is a separate column on the spreadsheet, the images arranged as rows of chronological order, with different parameters occupying different sheets of the spreadsheet workbook.
- Multiply the measurements Mean_Mac_Children_pHr_Count by Count_Mac, to generate the parameter Number of spots per image. Calculate the mean Count_Mac for each Beacon. Divide the Number of spots per image by the mean Count_Mac for that beacon, generating the parameter Number of spots per cell.
NOTE: Step 6.1.26 corrects any erroneous fluctuations that can occur in the iPSC-macrophage count (Count_Mac), by normalizing the data to the average iPSC-macrophage count across all of the timepoints of a Beacon. - Assign the time since phagocytosis commenced (in min) to each image row.
- Generate means and standard deviation for replicate wells/beacons. Graph the Number of spots per cell (y-axis) against time (x-axis) to visualize the rate of phagocytosis.
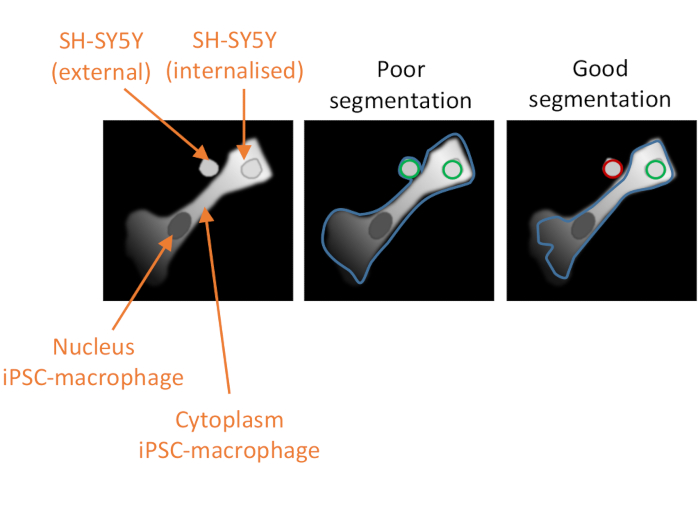
Figure 2: Cell segmentation in the high-content phagocytosis analysis. Illustration to demonstrate good versus poor segmentation of an iPSC-macrophage in close proximity to a non-phagocytosed SH-SY5Y, with a second SH-SY5Y fully phagocytosed. With both cell types shown in grey, the iPSC-macrophage cell border delineated by the computer analysis is outlined (blue). SH-SY5Ys that are counted as phagocytosis events are outlined green or outlined red if excluded from the analysis. The image in the middle shows poor segmentation; the iPSC-macrophage has sub-optimal delineation that includes the non-phagocytosed SH-SY5Y within the cell border, which will be counted as a phagocytosis event. The image on the right shows good segmentation due to more stringent parameters defining the iPSC-macrophage cell border, which led to the non-phagocytosed SH-SY5Y being correctly excluded from the analysis. Please click here to view a larger version of this figure.
- Analysis of phagocytosis images obtained with high-content microscope
- Log in to the recommended image-processing software (see Table of Materials).
- Select the screen name folder, and the imaging run sub-folder from the left-hand menu. Click on the Image Analysis icon (a screen with a magnifying glass). Select a representative well on the plate layout for setting up the analysis pipeline.
- The first analysis building block is input image. Leave the default settings for stack processing (Individual Planes) and flatfield correction (None). Click on the + sign at the top right corner of the block, to add the next building block, and select Find Nuclei.
- In Find Nuclei, set the channel as DAPI, the ROI population as None, the method of segmentation as C. The method box contains a drop-down menu that allows the parameter to be optimized, with settings for the common threshold (i.e., 0.40) and area (i.e., >30 µm2). Name the output population "Nuclei". Add the next building block by clicking on the + symbol and select Find Cytoplasm.
- In Find Cytoplasm, set the channel as Alexa 647 and the method as B. The method box contains a drop-down menu that allows the parameter to be optimized, with settings for the common threshold (i.e., 0.45) and the individual threshold (i.e., 0.20). Add the next building block by clicking on the + symbol and select Select Population.
NOTE: It is critical to properly optimize the cytoplasm segmentation so that it excludes any adjacent SH-SY5Ys that have not been phagocytosed but does not exclude phagocytosed cargo (see Figure 2). - In Select Population, keep the default settings, which will be population Nuclei, method Common Filters, a tick for Remove Border Objects, and the output population named "Nuclei Selected". Add the next building block by clicking on the + symbol and select Calculate Morphology Properties.
- In Calculate Morphology Properties, set the population to Nuclei Selected, the region to Cell, the method to Standard. In the drop-down menu, ensure that area and roundness are selected (µm2). Name the output population "Morphology Cell". Add the next building block by clicking on the + symbol and select Select Population.
- In Select Population (2), choose the population Nuclei Selected, and the method Filter by Properties. In the drop-down box under Filter F1, select Morphology Cell Area [µm2]. Choose > from the drop-down box to the right, and type 160 in the box to the right of that. Name the output population "Nuclei Selected 2". Add the next building block by clicking on the + symbol and select Find Spots.
NOTE: This step excludes any improperly segmented cells, and any dead cells from further analysis. It may be necessary to optimize by increasing or decreasing the cut-off size. - In Find Spots, select the channel Alexa 568, the ROI population Nuclei Selected 2, the ROI region Cell, method B, and name the output population "Spots". The method can be optimized, if necessary, using the drop-down menu, with settings for detection sensitivity (i.e., 0.20) and splitting sensitivity (i.e., 0.400). Add the next building block by clicking on the + symbol and select Calculate Morphology Properties.
- In Calculate Morphology Properties (2), select the population Spots, region Spot, and method Standard. In the drop-down menu, ensure that area and roundness are selected (µm2). Name the output properties "Morphology Spot". Add the next building block by clicking on the + symbol and select Select Population.
- In Select Population (3), select the population Spots and the method Filter by Properties. In the drop-down boxes under Filter F1, select Spot Area [px2], >, 20. In the drop-down boxes under Filter F2, select Spot Area [px2], <, 2500. In the drop-down boxes under Filter F3, select Morphology Spot Roundness, >, 0.6. In the drop-down boxes under Filter F4, select Spot to Region Intensity, >, 2.5. Name the output population "Spots Selected". Add the next building block by clicking on the + symbol and select Select Population.
NOTE: The automated spot selection will have segmented many tiny fluorescent specks that result from autofluorescent bodies within the iPSC-macrophages. This step aims to filter out autofluorescent bodies by applying stringent cut-offs to the area, roundness, and intensity of the spots, and may require some optimization. - In Select Population (4), select the population Nuclei Selected 2 and the method Filter by Properties. In the drop-down boxes under Filter F1, select Number of Spots, >, 0.5. Name the output population "Spot Positive Cells". Add the next building block by clicking on the + symbol and select Define Results.
- In Define Results, select the first method as List of Outputs. The default setting is for the number of objects to be calculated for each population. Click on the drop-down menu for Population: Nuclei Selected 2 and ensure that Number of Objects is ticked, and in the Apply to All drop-down menu select ALL. For the population Spot Positive Cell, ensure that Number of Objects is ticked. For the other populations, it is not necessary to report any parameters. Select the second method as Formula Output, and type the formula (a/b)*100. Choose as variable A Spot Positive Cell- Number of Objects, and as variable B choose Nuclei Selected 2- Number of Objects. Name the output as "Spot Positive Cells (%)".
- Save the pipeline: click on the icon Save Analysis to Disk (a floppy disk with down arrow).
- Click on the icon Batch Analysis (a funnel and cogs symbol along the top of the screen). From the experimental folders on the left, select the raw data file, which should update the number of selected measurements to 1. In the Analysis options region, click on the drop-down menu for Method, and select Existing Analysis. Click on the … symbol next to Script File, and browse for the saved analysis file (suffixed .aas). Then, click on the green arrow next to Start Analysis. The analysis progress can be monitored by clicking on Job Status (in the upper-right-hand corner of the screen).
- Once the analysis is complete, click on the tab Export, choose the experiment folder, and select a destination folder. Leave the default settings, which export data but not TIFF images, and start the export.
- Open the downloaded file as a spreadsheet in an appropriate spreadsheet software. The wells are arranged in rows and the parameters in columns. Select the data in columns labeled Spot Positive Cells (%), Nuclei Selected 2 – Number of Spots – Mean per Well, and Nuclei Selected 2 – Total Spot Area – Mean per Well, and copy these to fresh spreadsheets for each parameter. Calculate the parameter mean for replicate wells of each condition, and graph as appropriate.
7. Quality control assay for homogeneity of fixed SH-SY5Ys
- Collect an aliquot of live SH-SY5Ys from step 2.2 and resuspend in the annexin binding buffer from a kit for annexin V-FITC staining (see Table of Materials) at a concentration of approximately 200,000 cells per mL.
- Collect an aliquot of fixed SH-SY5Ys from step 2.4 and resuspend in annexin binding buffer at a concentration of approximately 200,000 cells per mL.
- Prepare two test tubes with 5 µL of annexin V-FITC and 5 µL of propidium iodide (see Table of Materials). Add 500 µL of live SH-SY5Ys to one tube, and 500 µL of fixed SH-SY5Ys to the other.
- Prepare three control tubes: one with 5 µL of annexin V-FITC, one with 5 µL of propidium iodide, and one tube empty. Mix together a 1:1 ratio of live and fixed SH-SY5Ys and add 500 µL of this to each control tube.
- Mix tubes gently by pipetting. Incubate at room temperature for 10 min, protected from the light.
- Immediately measure on a flow cytometer(Ex = 488 nm; Em = 530 nm) using FITC signal detector (usually FL1) for annexin V-FITC, and the phycoerythrin emission signal detector (usually FL2) for propidium iodide.
- Use any flow cytometry analysis software to display dot plots of FITC vs PI signal and use a rectangular gating tool to select the double-negative population. Within the double-negative population, display FSC vs SSC and use a polygonal gating tool to create an exclusion gate around the population with very low FSC and SSC, which is classified as debris and therefore excluded from further analysis. Display the remaining events as FITC vs PI signal and use the single-stained and unstained controls to set a quadrant gate for FITC-/PI-, FITC+/PI-, FITC -/PI+, and FITC+/PI+ events.
NOTE: Avoid rough handling, vortexing, or long incubations with live SH-SY5Ys, which could artificially induce phosphatidylserine display. Proceed to flow cytometry without delay. A desirable outcome is that the proportion of FITC-/PI- events is <5% in the fixed SH-SY5Ys. Representative results are shown in Supplementary Figure S1.
Live-cell time-lapse imaging was performed using the previously outlined protocol, with wild-type iPSC-macrophages seeded at 20,000 cells per well. Different amounts of SH-SY5Ys were applied (10,000-30,000 per well as estimated from the cell count in step 3.1), and the phagocytosis inhibitor cytochalasin D was pre-incubated (1 h) with some wells, acting as a control to inhibit phagocytosis for each amount of SH-SY5Ys. Imaging commenced 40 min after the addition of SH-SY5Ys and images were captured at 5 min intervals for the next 3 h (data includes the initial 40 min delay). A representative time-lapse video is included in the Supplementary Data, and analyzed quantitative data shown in Figure 3. With the amount of 10,000 SH-SY5Ys per well, the number of phagocytosed particles (spots) per cell increased linearly with time, and was inhibited by approximately 50% by cytochalasin D. The inhibition by cytochalasin D was weaker than anticipated, most likely caused by insufficient technical or biological replicates, as only one well per condition was imaged with three image fields. With higher amounts of SH-SY5Ys per well (20,000 and 30,000), phagocytosis exhibited poor linearity, likely due to poor segmentation of iPSC-macrophages and SH-SY5Ys in a more crowded field of view.
Fixed-cell high-content imaging was performed using the previously-outlined protocol, with wild-type iPSC-macrophages at 20,000 cells per well, several different amounts of SH-SY5Ys (10,000-80,000 per well), and the assay plate was fixed and imaged after 5 h. A representative image of phagocytosis is presented in Figure 4A, and the analyzed data shown in Figure 4B17. Increasing the amount of SH-SY5Ys resulted in a higher number of phagocytosed particles (spots) per cell; however, a doubling of SH-SY5Y quantity only leads to a 1.5x increase in number of spots per cell. This indicates that the amounts tested are not rate-limiting to phagocytosis. Subsequently the high-content imaging phagocytosis assay was validated using several inhibitors of phagocytosis (Figure 4C)17. The actin polymerization inhibitors cytochalasin D and jasplakinolide significantly inhibited phagocytosis by 91% and 90%, respectively, when pre-incubated for 1 h prior to phagocytosis. The robust Z' of the assay when cytochalasin D or jasplakinolide are used as negative controls is calculated as 0.7 and 0.8, respectively20. The lysosome acidification inhibitor bafilomycin A1 significantly reduced phagocytosis by 31%, when incubated 1 h prior to phagocytosis. The weaker effect of the lysosome acidification inhibitor versus actin inhibitors suggest that detection of the internalized cargo may not require full acidification of the phagosome. Recombinant annexin V was used as a control to specifically block phosphatidylserine exposed on the surface of SH-SY5Ys, preventing phagocytic receptors from accessing the ligand, an important "eat-me" signal. Addition of recombinant annexin V significantly reduced phagocytosis by 30%, when added to wells immediately before SH-SY5Y addition. Fixed SH-SY5Ys were confirmed to expose phosphatidylserine, using a fluorescent annexin V probe, whereas live SH-SY5Ys were negative for annexin V staining (Figure 4D).
The microglial phagocytosis receptor TREM2 has previously been shown to be important for phagocytosis of apoptotic neurons21. The R47H mutation of TREM2 is a risk gene for late-onset of Alzheimer's disease, and is hypothesized to reduce ligand binding of TREM223. With the aim of assessing phagocytic function of R47H TREM2 and TREM2 KO, the fixed-cell high-content phagocytosis assay was performed using isogenic iPSC-macrophage lines with WT/R47H/KO TREM217. Several lengths of phagocytosis duration from 1 to 5 h were tested, using a staggered addition of phagocytic cargo (40,000 SH-SY5Ys). The resulting signal increases linearly to 4 h, leveling off slightly at 5 h (Figure 5)17. Reduced phagocytosis rate and capacity (% spot positive cells) was evident in the TREM2 KO compared to WT, whereas the R47H TREM2 mutant did not show altered phagocytosis. The phagocytic defect in TREM2 KO cells is not phenocopied by the R47H TREM2 mutation, seemingly because the TREM2 function is sufficient to support normal phagocytosis.
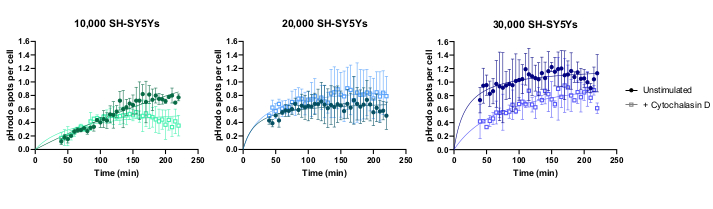
Figure 3: Example data for live-cell time-lapse phagocytosis assay. Uptake of dead SH-SY5Ys by wild-type iPSC-macrophages BIONi010-C (ECACC ID: 66540023) imaged at 5 min intervals for 3 h. Times displayed on the graph are from initiation of phagocytosis, including the first 40 min without measurement. The average number of spots per cell from three replicate wells are plotted. Phagocytosis of 10,000 SH-SY5Ys is inhibited with 10 µM cytochalasin D with 1 h pre-treatment, whereas higher amounts of SH-SY5Ys (20,000 and 30,000) have suboptimal quantification of phagocytosis. Mean ± standard deviation (SD), N = 1 experiment. Please click here to view a larger version of this figure.
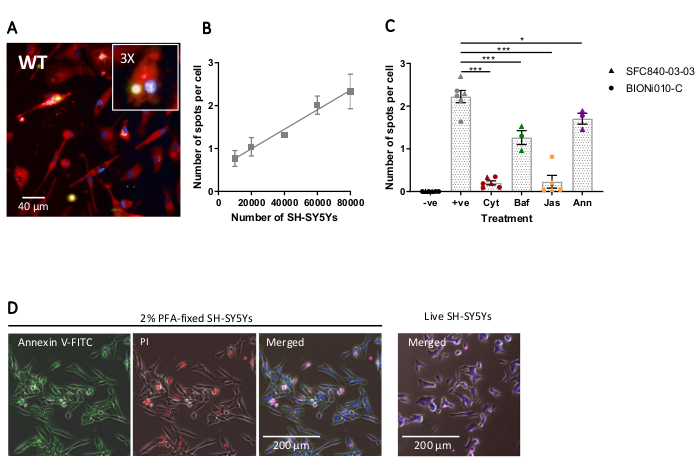
Figure 4: Optimization and validation of fixed-cell high-content phagocytosis assay. (A) Representative high-content microscopy image of SH-SY5Ys phagocytosed by wild-type iPSC-macrophages BIONi010-C (ECACC ID: 66540023). A 3 h time-point with 40,000 SH-SY5Ys is shown. Fluorescence channels are merged, with iPSC-macrophage stain shown as red, nuclei as blue, and SH-SY5Ys as yellow. The inset panel is a section of the image magnified 3x. (B) The number of spots per cell of phagocytosed dead SH-SY5Ys after 5 h, using different amounts of cargo addition to wild-type iPSC-macrophages. Mean ± standard error of the mean (SEM), for N = 3 harvests. (C) Phagocytosis (3 h) is inhibited with 10 µM cytochalasin D (Cyt), 1 µM bafilomycin A1 (Baf), 1 µM jasplakinolide (with 1 h pre-treatment; Jas), and 13 µg/mL recombinant annexin V (added simultaneously to the dead SH-SY5Ys; Ann). iPSC-macrophages with no SH-SY5Ys added were used as a negative control (-ve), and the positive (+ve) control is untreated iPSC-macrophages with SH-SY5Ys added. Data was normalized to the mean for the experiment repeat. Means ± SEM, for N = 3-6 harvests and with two wild-type cell lines (SFC840-03-03, the characterization of this line is described in (Fernandes et al.21 and BIONi010-C). 1-way ANOVA with Dunnett's post-hoc test, comparisons to untreated cells. *p < 0.05, ***p < 0.001. (D) Freshly fixed SH-SY5Ys stain uniformly for phosphatidylserine display (annexin V-FITC) and have limited cell permeability (propidium iodide). Live SH-SY5Ys do not stain for annexin V-FITC or propidium iodide, except for focal staining present on the few dead cells in culture. Figures are reproduced with permission from Alzheimer's Research & Therapy17. Please click here to view a larger version of this figure.
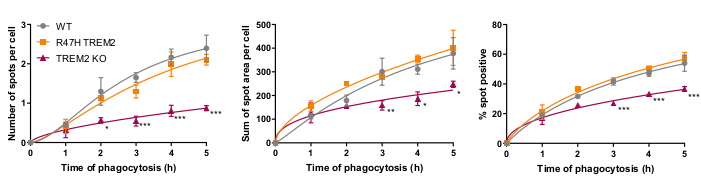
Figure 5: Phagocytosis is reduced in TREM2 KO but not in R47H TREM2 iPSC-macrophages. High-content phagocytosis assay performed with 40,000 SH-SY5Ys per well with staggered additions. Means were quantified for the parameters: number of spots per cell, sum of spot areas (µm2) per cell, and percentage of cells containing phagocytosed particles per field. Data was normalized to mean for each genotype per experiment. Mean ± SEM, for N = 3 harvests. Repeated-measures 2-way ANOVA, Dunnett's post-hoc test, pairwise comparisons to the WT for each time: *p < 0.05, **p < 0.01, ***p < 0.001. Figures are reproduced with permission from Alzheimer's Research & Therapy17. Please click here to view a larger version of this figure.
Supplementary Figure S1: Example QC for SH-SY5Ys preparation. Dissociated SH-SY5Ys were fixed for 10 min with 0% (live cells), 1%, and 2% of paraformaldehyde (PFA), then washed. Cells were stained with annexin V-FITC and propidium iodide (PI) and immediately measured by flow cytometry. Color density dot plots were created in flow cytometry analysis software, using the single-stained and unstained controls to place a quadrant gate. Quadrants are annotated with the percentage of events within that quadrant. Live cells are mainly in Q4, and fixed cells are mainly in Q2. Q1 = annexin V-/PI-, Q2 = annexin V+/PI+, Q3 = annexin V+/PI-, Q4 = annexin V-/PI- (live cells). Please click here to download this File.
Supplementary Video: Live-cell time-lapse phagocytosis. Representative time-lapse video of SH-SY5Ys phagocytosed by wild-type iPSC-macrophages BIONi010-C (ECACC ID: 66540023). Images were taken every 5 min for 3 h. Video is cropped and runs at 3 frames per second, showing the last 1.5 h of the assay. The acid-sensitive dye-stained SH-SY5Ys are shown in red, signal intensity increasing with phagosome acidification. Cell nuclei stained with Hoechst 33342 are shown in blue. Please click here to download this Video.
