Protocol Validation
The iPSC-derived microglia were generated from seven iPSC lines over three different rounds of differentiation. Control iPSC lines ML27, ML56, ML292, and ML364 and schizophrenia iPSC lines ML40, ML141, and ML250 were utilized. Characterization of these iPSC lines have been described previously51. These iPSC-derived microglia were validated using ICC and qPCR. Microglia generated from the adapted protocol exhibited typical ramified microglial morphology (Figure 1C), and expressed microglial markers CD11c, TMEM119, and IBA1, as examined by immunocytochemistry (Figure 1D,E). Cells with nuclei expressing microglial markers CD11c, P2RY12, and IBA1 were quantified. CD11c, P2RY12, IBA1 and TMEM119 were present in 63%, 60%, 65%, and 44% of the cells respectively, which is consistent with data described in the original differentiation protocol paper49. These experiments were performed with iPSC lines ML27, ML40, ML141, and ML250. Expression of specific genes was examined using qPCR to confirm the expression of microglial genes AIF1, CX3CR1, ITGAM, ITGAX, P2RY12, and TMEM119 (Figure 1F). This data was obtained from iPSC-derived microglia from two lines and normalized to an iPSC line. The SYBR Green real-time PCR protocol was used.
Dendritic Spines
Cortical neurons and microglia in co-culture were visualized using confocal microscopy (Figure 2A). Dendritic spines were quantified in the co-cultured cortical neurons (Figure 2B). Co-cultures were analyzed to determine the proportion of microglia and cortical neurons by marker analysis using P2RY12 and MAP2 to identify microglia and neurons respectively. In these co-cultures, 32.5% of the cells were positive for P2RY12 and 37.7% of the cells were positive for MAP2. Cortical neurons co-cultured with microglia treated with IFN-γ exhibited no significant differences in spine count, spine length, and neurite length when compared to cortical neurons co-cultured with untreated microglia (Figure 2D). This data was collected from the four control iPSC lines ML27, ML56, ML292, ML364, with two separate wells per experimental condition and ten images obtained from each well.
Calcium Imaging
Neurons co-cultured with microglia were stained with calcium fluorescence indicator in order to examine differences in neuronal firing with stimulation from glutamate with and without IFN-γ induced microglial activation (Figure 2E). Cortical neurons co-cultured with microglia treated with IFN-γ showed significant reduction in fluorescence intensity after stimulation with glutamate compared to cortical neurons co-cultured with untreated microglia (Figure 2F). This data was collected for three healthy control iPSC lines ML27, ML56, and ML292, with two wells per experimental condition and five images obtained from each well.
Supplementary Figure 1 further validates antibodies and staining protocol.
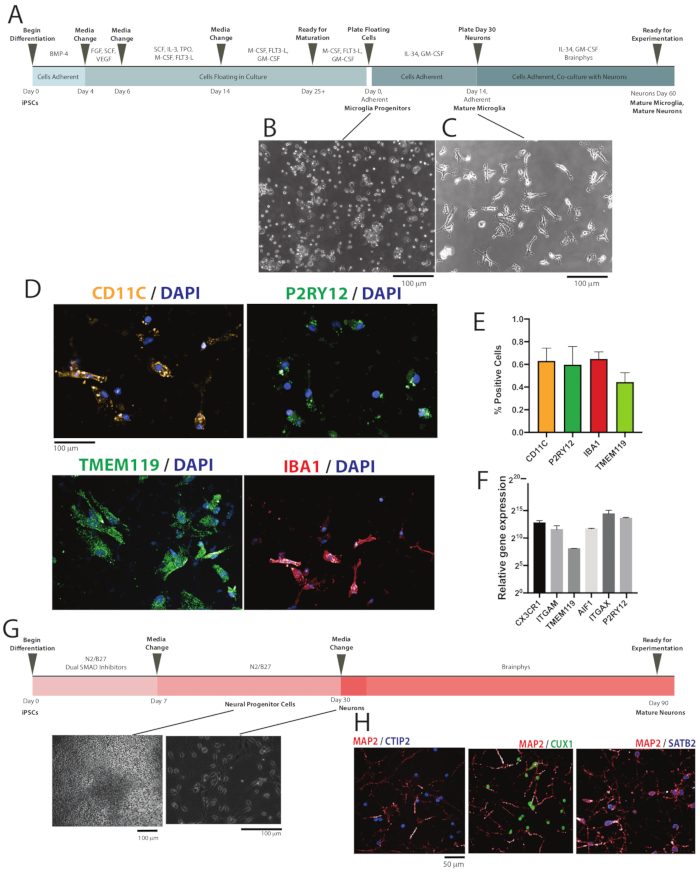
Figure 1: Differentiation and validation of iPSC-derived microglia. (A) Schematic depiction of microglial differentiation from iPSCs through microglial maturity and co-culture with cortical neurons. (B) Representative image of microglial progenitor cells after re-plating following day 25 of differentiation at 10x magnification. (C) Representative image of microglia in monoculture at day 14 at 10x magnification. (D) Immunocytochemistry staining of CD11c, P2RY12, IBA1, and TMEM119 to confirm expression of microglial markers, shown at 20x magnification. (E) Percentage of cells positively stained for microglial markers CD11c, P2RY12, IBA1, and TMEM119. (F) qPCR validation showing microglia exhibiting microglial-signature genes including AIF1, CX3CR1, ITGAM, ITGAX, P2RY12, and TMEM119. (G) Schematic depicting cortical neuron differentiation from iPSCs. (H) Immunocytochemistry staining of CTIP2, CUX1, SATB2, and MAP2 to confirm generation of cortical neurons, shown at 63X magnification. Please click here to view a larger version of this figure.
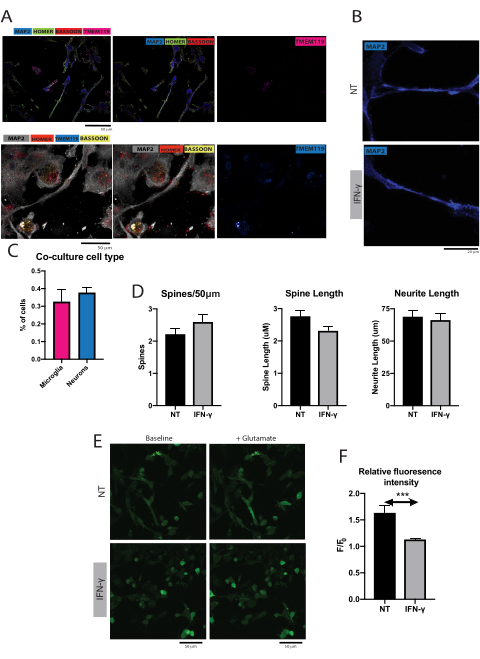
Figure 2. Functional changes in microglia and neuron co-cultures with and without interferon-gamma treatment. (A) Representative image of microglia and cortical neuron co-culture, with neurons stained for MAP2, pre-synaptic marker bassoon and post-synaptic marker homer, and microglia stained for TMEM119. (B) Representative image of dendritic spines in microglia/neuron co-cultures, with and without IFN-γ treatment. Scale bar = 50 μm. (C) Percentage of cells positive for P2RY12 or MAP2 in the co-cultures (mean + SEM). (D) Cortical neurons co-cultured with microglia treated with IFN-γ showed no significant differences when compared to untreated microglia in terms of spine count (Mann-Whitney test, P > 0.05, mean + SEM), spine length (Mann-Whitney test, P > 0.05, mean + SEM), or neurite length (unpaired t test, P > 0.05, mean + SEM). (E) Representative images of cortical neurons co-cultured with microglia in the presence of the calcium indicator Fluo-4AM, before and after glutamate stimulation and with and without IFN-γ treatment. (F) Cortical neurons co-cultured in the presence of microglia treated with IFN-γ had significantly lower fluorescence intensity with glutamate stimulation when compared to neurons co-cultured with untreated microglia (Mann-Whitney test, P = 0.0003, mean + SEM). Please click here to view a larger version of this figure.
| Protocol | Supplements used | Sorting stage | Length of Differentiation | Protein markers expiressed | Characterization | |||
| Abud et al. | FGF, BMP4, Activin A, LiCl, FGF, VEGF, TPO, SCF, IL-3, IL-6, M-CSF, IL-34, TGFb1, insulin, CD200, CX3CL1 | At day 10, isolate CD34+ cells | 38 days | CD45, CX3CR1, ITGB5, MERTK, PROS1, TGFbR1, P2RY12, TREM2 | Cytospin/Giemsa staining, transcriptomic profiling, RNA seq, flow cytometry, RT-QPCR, cell type analysis, flow cytometry, motility assay, inflamation response assay, phagocytosis assay, transplantation | |||
| Douvaras et al. | BMP4, FGF, SCF, VEGF, IL-3, TPO, M-CSF, Flt-3L, GM-CSF, IL-34 | At day 25, for CD14+/ CX3CR1+ cells | 40 days | CD11b, CD11c, CX3CR1, IBA1, P2RY12, TMEM119 | ICC, RT-QPCR, cell type analysis, flow cytometry, RNA seq, calcium assay, motility assay | |||
| Haenseler et al. | BMP4, SCF, VEGF, M-CSF, IL-3, GM-CSF | None | 42 days | CD11b, CD14, CD45, IBA1, MERTK | ICC, RT-QPCR, cell type analysis, flow cytometry, motility assay, inflamation response assay | |||
| Mcquade et al. | IL-34, TGF-β1, M-CSF, CLxCL1, CD200 | None | 38 days | P2RY12, TMEM119 | ICC, RNA seq, phagocytosis assay, transplantation assay | |||
| Muffat et al. | M-CSF, IL-34 | None | 74 days | CD11B, IBA1, P2RY12 TMEM119 | ICC, RNA seq, flow cytometry, cell size comparison Endotoxin response, motility assay | |||
Table 1: Overview of current protocols for differentiation of iPSCs to microglial cells.
| Day | Medium | Cytokines | Concentration |
| 0-3 | Stem Cell Medium | BMP-4 | 80ng/mL |
| 4-5 | Hematopoietic Medium | FGF | 25ng/mL |
| SCF | 100ng/mL | ||
| VEGF | 80ng/mL | ||
| 6-13 | Hematopoietic Medium | SCF | 50ng/mL |
| IL-3 | 50ng/mL | ||
| TPO | 5ng/mL | ||
| M-SCF | 50ng/mL | ||
| Flt3-Ligand | 50ng/mL | ||
| 14-25+ | Hematopoietic Medium | M-SCF | 50ng/mL |
| Flt3-Ligand | 50ng/mL | ||
| GM-SCF | 50ng/mL | ||
| Adherent | RPMI 1640 | GM-SCF | 25ng/mL |
| IL-34 | 100ng/mL |
Table 2: Overview of media used for microglial differentiation, listed with the concentration of cytokines used.
| Protocol | Day 0-3 Medium | Feeding method | Sorting | Supplements |
| Douvaras et al. | Custom medium; medium without Lithium Chloride, GABA, Pipecolic Acid, bFGF and TGFβ1 supplemented with 80ng/mL BMP4 | Every four days, cells pelleted and resuspended in fresh medium | Isolation of CD14+ or CD14+CX3CR1+ progenitors via FACS sorting | |
| This protocol | iPSC medium supplemented with 80ng/mL BMP4 | Every four days fresh medium added on top of existing medium | None | Rock inhibitor (10μM) used after centrifugation |
Table 3: Brief overview of microglial differentiation protocol from Douvaras et al.49 and adaptations made in this study.
| Protocol | Neural Maintenance Medium | Neural Induction Medium | Supplements | Notable Differences | ||
| Shi et al. 201250 | N2/B27 medium. N2 medium: Basal medium with 1% N-2 supplement, 1% 200mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl solution, 1% pen/strep, 5μg/mL insulin, 1mM ;-glutamine, 100μM non-essential amino acids, 100μM 2-mercaptoethanol. B27 medium: DMEM/F12 supplemented with 2% B-27 supplement, 200mM L-glutamine, 1% pen/strep. | N2/B27 medium supplemented with 10μM SB431542 and 100ng/mL noggin OR 10μM SB431542 and 1μM dorsomorphin. | 20ng/mL FGF2 upon appearance of rosettes | Use of Insulin, NEAA, 2-mercaptoethanol | ||
| This protocol | N2/B27 medium. N2 medium: Basal medium with 1% N-2 supplement, 1% 200mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl solution, 1% pen/strep. B27 medium: DMEM/F12 supplemented with 2% B-27 supplement, 1% 200mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl solution, 1% pen/strep. | N2/B27 medium supplemented with 10μM SB431542, 1μM dorsomorphin, 100nM LDN193189. | Use of 200mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl solution in B27 medium, use of 100nM LDN193189 in Neural Induction Medium | |||
Table 4: Brief overview of neuronal differentiation protocol from Shi et al.50 and adaptations made in this study.
Supplementary Figure 1: (A) Control iPSCs stained Microglia specific antibodies CD11c, P2RY12 and (B) TMEM119 and Iba1 to ensure these antibodies do not stain non-microglia cells. Control iPSC line did not exhibit any positive staining for these markers. (C) Co-cultures analyzed by cell type visible by specific line. Co-cultures were stained with a neuronal marker MAP2 or microglial marker P2RY12. Please click here to download this File.
Supplementary Table 1: List of primary antibodies used in this protocol and their optimal dilutions. Please click here to download this Table.
Supplementary Table 2: List of primers used for RT PCR experiments with forward and reverse sequences. Please click here to download this Table.
