Optimization of the Single Reporter Serological Assay.
The coupling strategy for all SARS-CoV-2 antigens is described in Cameron, et al., 20201 and in step 2.1 above. The strategy for coupling confirmation is described in step 2.3 above. For an efficient coupling, the median fluorescence intensity (MFI) values should be significantly higher than the no serum/no antibody negative control reaction(s) and should demonstrate a dose response of decreasing MFI signal with decreasing amounts of detection reagent. MFIs of approximately 1,000 MFI units or greater over background MFI generally indicate efficient protein coupling and are dependent on the concentration and specificity of the detection reagent used. Using this method, confirmation of S, RBD, and Nc antigen coupling were tested for two lots of beads (Figure 2). The MFI signals for all antigens were significantly higher than the background MFI and showed a dose response with titration of the detection reagents. The data also showed good reproducibility of MFI levels between the two different lots of coupled beads.
Optimization of the single reporter assay was done as described in Cameron, et al., 20201. Each antigen was tested at different coupling concentrations with different dilutions of several serum samples representing high, medium, low, and negative samples. In addition, because the assay measures IgG titers, an IgG-stripped serum was used as an additional negative sample. Results of a representative sample dilution series with different antigen coupling levels are shown in Figure 3. This initial dilution series was tested with a fixed concentration of detection reagents to get a potential range of representative antigen-specific and background MFI levels. From these data, additional sample dilution ranges and antigen coupling levels were determined and tested further with additional samples (data not shown).
Next, the concentration of the detection reagent was optimized for use with the different serum dilutions and antigen coupling levels. Representative data using 6 positive and 2 negative samples are shown in Figure 4. Following additional testing with several hundred negative pre-COVID-19 serum samples, the final optimal antigen coupling levels and detection regent concentrations were selected for the final assay protocol, as described in section 5.
Conversion to a single reporter neutralization assay
Conversion of the single reporter serological assay to a neutralization assay was achieved by adding an incubation step using ACE2 (as a competing reagent) with the beads prior to adding the serum sample. By titrating the amount of ACE2 added, inhibition of IgG binding to the S and RBD antigens was observed, as shown in Figure 5. While the 11 patient sera had different titers of IgG, each showed a significant decrease in MFI signal with increasing ACE2 concentration. As expected, ACE2 only impacts the MFI signals of S and RBD, but not the Nc antigen.
The percent residual MFI signal relative to 0 µg/mL ACE2 for all samples is shown in Figure 6. For most samples, a signal loss >30% was observed for S and RBD with increasing ACE2 concentration. As expected, there was no effect of ACE2 on the Nc signal.
Conversion to a dual reporter IgG and IgM serological assay
For the dual reporter IgG and IgM assay, the standard single reporter assay conditions were used to test different antibody and reporter combinations for the two reporter channels. The RP1 channel is similar to previous flow analysis instruments with detection of "orange" fluorescence for reporter dyes such as PE. The second channel, RP2, employs a violet excitation laser that can be used for detecting the "blue" fluorescence of dyes excited at 402 nm with emission spectra peaks in the 421-441 nm range.
Several different anti-human IgG and IgM antibody combinations were tested at various concentrations with the standard 2,000-fold sample dilution and incubation conditions detailed in section 6. An initial test of anti-IgM conjugated to the DyLight 405 reporter dye (DL 405; excitation at 400 nm and emission at 420 nm) for the RP2 channel was performed using a set of 11 PCR-positive samples with DFSO ranging from 5 to >60 days, and an IgG-stripped serum sample. This detection reagent did not produce a high signal in the RP2 channel as shown in Figure 7A,B,C. For samples with the high IgM RP2 MFI, the highest signals were seen for RBD and Nc (Figure 7B,C). While IgM titers should be elevated at this time in some samples, the observed MFI levels did not exceed 140 MFI units with the IgM detection reagent. Furthermore, the control bead for IgM lacked a significant dynamic range for MFI when using DL 405 conjugated to anti-IgM compared to a PE-labeled anti-IgM RP1 reporter used at the same concentrations (Figure 7D).
Because a suitable RP2 reporter-labeled detection reagent for IgM was not available, the original biotin anti-IgG with SASB was tested for use in the RP2 channel. The signal for IgG in the RP2 channel with SASB did not have the same dynamic range as with SAPE, but it still had a suitable range for measuring IgG titers for S, RBD, and Nc (Figure 8). These data led to testing different RP1 IgM and RP2 IgG detection reagent combinations, as described below.
Conversion to dual reporter neutralization assay
The dual reporter serological assay was converted to a neutralization assay by adding an incubation step with ACE2 and beads, prior to adding the serum sample. A 2-fold dilution series of ACE2 starting at 16 µg/ml was used, and then tested with sets of 11 samples and stripped sera as described above. In addition, 3 different combinations of RP1 IgM and RP2 IgG detection antibody mixes were tested on a set of 11 samples and stripped serum controls. The final combinations and reagent concentrations determined to be optimal are detailed in Table 1.
For IgG detection, the biotin anti-human IgG/SASB used in the original single reporter assay was tested along with another anti-IgG conjugated to the BV reporter dye. While the BV conjugate had high RP2 MFI signals, the MFI signal intensity for IgG titer varied across the ACE2 titrations (Figure 9A,C,E). The biotin anti-human IgG/SASB combination had more consistent MFI signal decrease across the ACE2 titration compared to the BV conjugate, demonstrating a dose response, although the MFI levels were lower. The signal fluctuation by the BV conjugate across ACE2 concentrations also interfered with determining the percent inhibition by ACE2 across the range of IgG titers compared to the biotin anti-human IgG/SASB combination (Figure 9B,D,F). Samples with low MFI signals, such as the DFSO 3 sample, do not have high enough titers to evaluate the performance of these reagents or measure IgG neutralizing capacity.
For determining IgM titers in the RP1 channel, a PE-conjugated anti-IgM was compared to an anti-human IgM conjugated to the DyLight 549 reporter dye (DL 549; excitation at 562 nm and emission at 576 nm) at the concentrations listed in Table 1. Of these two reagents, the PE anti-IgM displayed higher MFIs than those generated by DL 549 anti-human IgM for the samples tested (Figure 9A,C,E). The IgM control bead measuring the addition of these reagents had different average MFIs of ≈17,000 for the PE anti-IgM and 2,723 for the DL 549 conjugate. Even with these differences, the impact of the various ACE2 concentrations on MFI was measurable with the DL 549 conjugate. For determining ACE2 percent inhibition of IgM binding, there was a slight but insignificant difference between the two IgM detection reagents (Figure 9B,D,F).
| Combination | Reporter | Final Concentration In Well (µg/mL) | |
| RP1 | RP2 | ||
| 1 | PE anti-IgM | 2 | – |
| Biotin-Ig | – | 0.62 | |
| SASB | – | 4 | |
| 2 | DyLight 549 Anti-Human IgM | 1 | – |
| Brilliant Violet 421 Anti-Human IgG | – | 1 | |
| 3 | DyLight 549 Anti-Human IgM | 1 | – |
| Biotin-Ig | – | 0.62 | |
| SASB | – | 4 | |
Table 1: List of RP1 and RP2 reporter dye combinations tested. Several different RP1 and RP2 reporter dyes were evaluated for measuring IgG and IgM titers. The three combinations shown above were tested at different RP1 detection modes and for optimization of the neutralization assay. For RP2 combinations using SASB, concentrations for the biotinylated detection antibody and SASB are shown. *Final Concentration represents the final concentration in the reaction well.
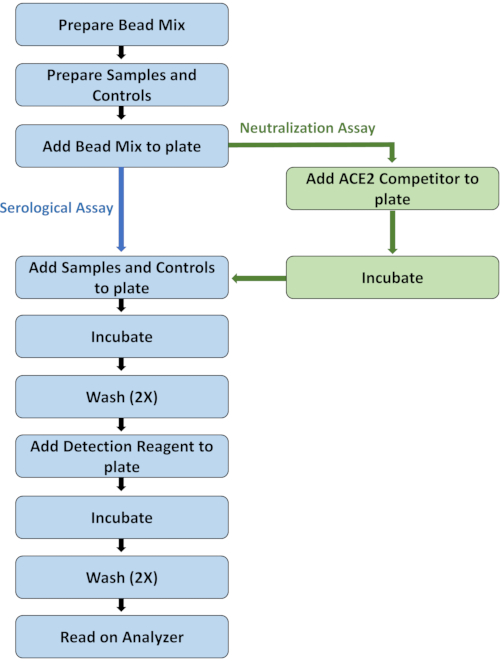
Figure 1: Flowchart highlighting main assay protocol steps. Please click here to view a larger version of this figure.
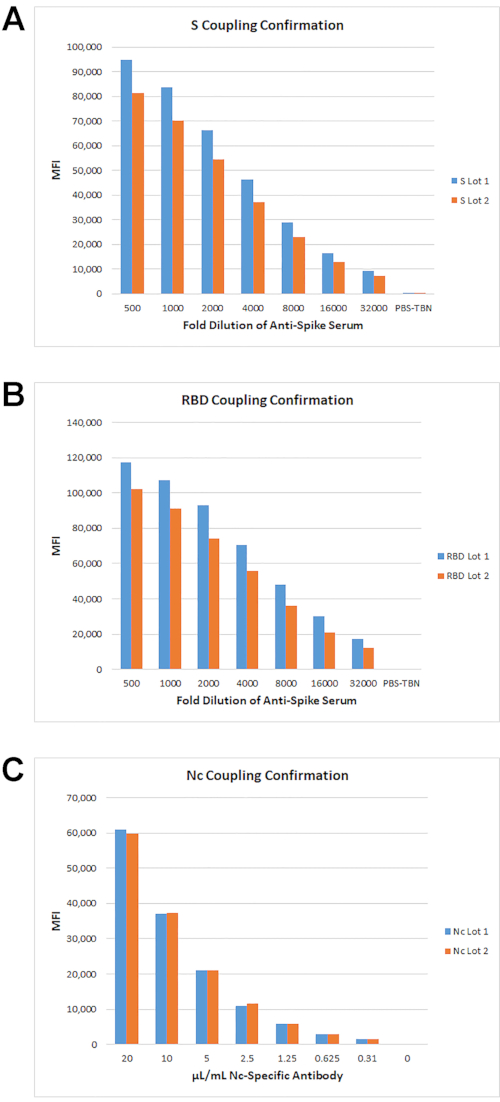
Figure 2: Confirmation of antigen coupling. Antigens were coupled at 100, 10 and 1 pmol/106 beads. For S and RBD antigens, rabbit anti-S sera was used and detected with PE anti-rabbit IgG. For Nc, a Protein G-purified rabbit polyclonal anti-Nc was used. The titration curves for beads coupled with 10 pmol S (A), 100 pmol RBD (B), and 100 pmol Nc (C) are shown. Please click here to view a larger version of this figure.
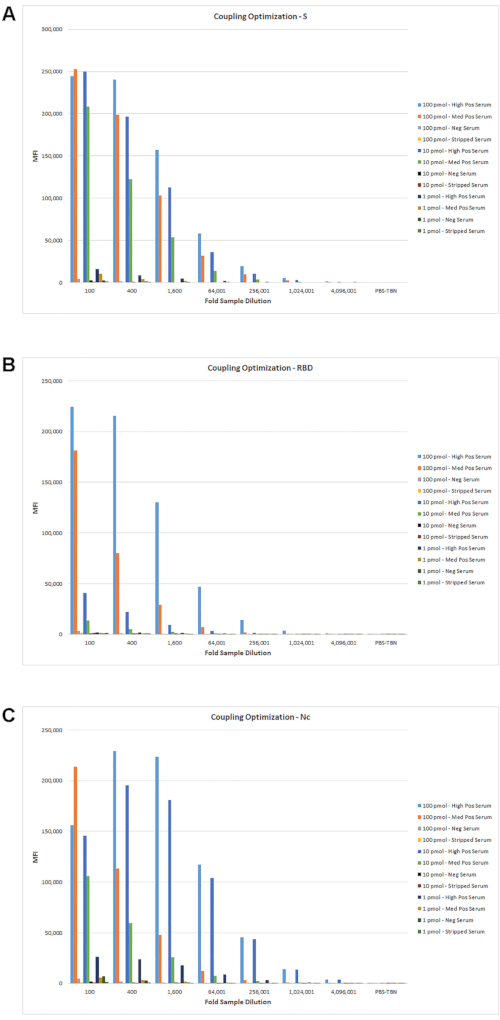
Figure 3: Optimization of serum dilution with different antigen coupling levels. Serum samples representing high, medium, low, and negative samples were tested with the different pmol antigen/106 bead couplings. A 4-fold serum titration was tested with a multiplex mix of antigen-coupled beads using the assay protocol described in section 5. The MFI values of the titration curves for S (A), RBD (B), and Nc (C) are shown. Please click here to view a larger version of this figure.
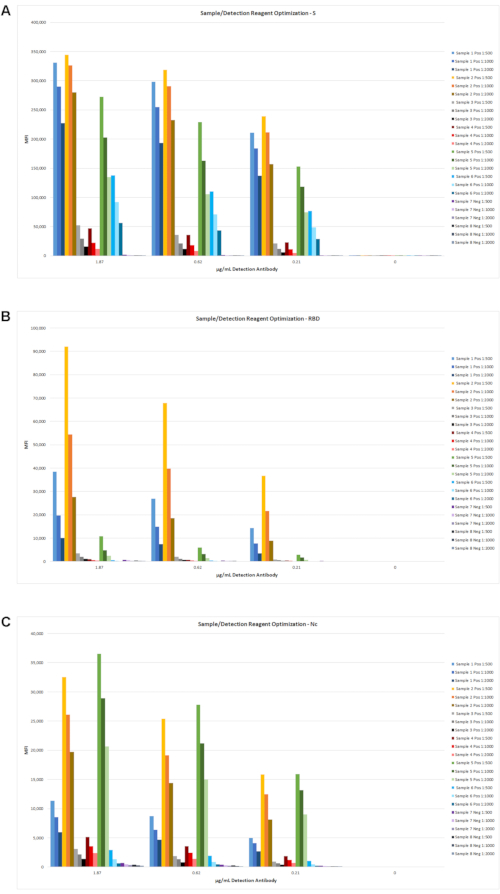
Figure 4: Determination of optimal serum dilution and detection reagent concentration. Eight serum samples, including negative patients (2 samples) and low- to high-positive (6 samples), were tested at additional serum dilutions with three different detection reagent concentrations. The MFI results are shown for S (A), RBD (B), and Nc (C). Based on these and other data (not shown), a sample dilution of 1:2,000 and a biotin detection antibody concentration of 0.62 µg/mL was selected. Please click here to view a larger version of this figure.
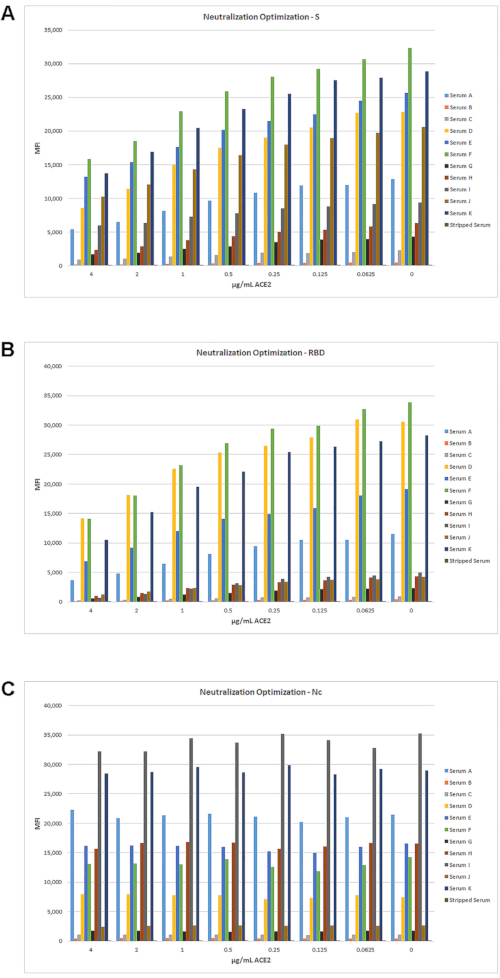
Figure 5: Optimization of single reporter neutralization assay. Modification of the single reporter serological assay to a neutralizing antibody detection assay was performed by adding an incubation step with ACE2 as a competitor, as described in Section 6. A set of 11 samples, ranging from low- to high-positive titer sera, and an IgG-striped serum sample were tested with a two-fold dilution series of ACE2 starting at 4 µg/mL, using standard assay conditions. Across this range of ACE2 concentrations, a decrease in MFI can be seen with increased ACE2 for S (A) and RBD (B), but not for Nc (C). Please click here to view a larger version of this figure.
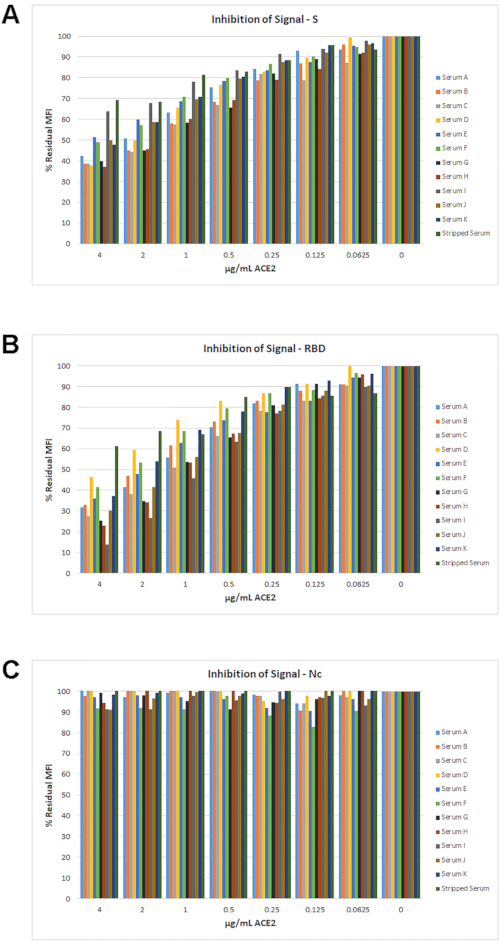
Figure 6: Percent inhibition of signal with ACE2. The percent residual signals for the set of 11 samples (from Figure 4) are shown. For each sample, regardless of its IgG titer, a maximum decrease of ≥30% MFI signal was achieved for S (A) and RBD (B) at the highest ACE2 concentration. For the Nc antigen (C) there was no significant inhibition of signal with any amount of ACE2 tested. Please click here to view a larger version of this figure.
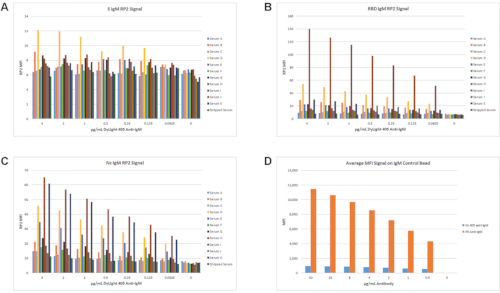
Figure 7: Test of DyLight 405 anti-IgM as RP2 IgM detection reagent. A set of 11 samples and IgG-stripped sera were used to test DL 405-conjugated anti-IgM as an RP2 detection reagent. The RP2 IgM MFI signals for S (A), RBD (B), and Nc (C) are shown. The highest MFI signal for any antigen with any sample was approximately 140 MFI units for RBD with sample H (B). Even the signal on the IgM control bead set was less than 1,000 MFI in the RP2 channel across the whole antibody titer range and did not show the increased dynamic range observed with the PE anti-IgM detection antibody in the RP1 channel (D). Please click here to view a larger version of this figure.
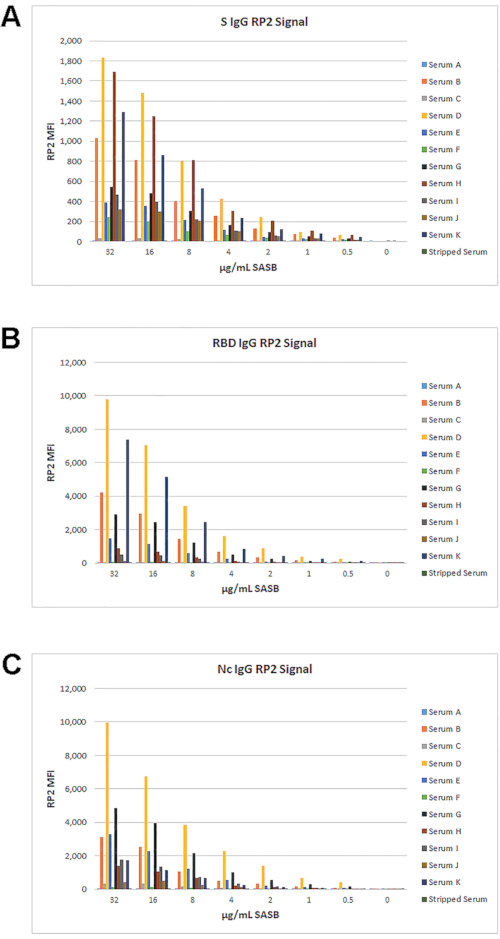
Figure 8: Optimization of SASB as RP2 reporter with Biotin anti-IgG. A set of 11 samples and IgG-stripped sera were used to test a dilution series of SASB with the standard 0.62 µg/mL biotin anti-IgG. The resulting IgG MFIs in the RP2 channel for S (A), RBD (B), and Nc (C) are shown. Please click here to view a larger version of this figure.

Figure 9: Comparison of three different combinations of RP1 IgM and RP1 IgG detection mixes. Three different detection antibody mixes were tested on 11 samples and IgG stripped sera. The combinations and final concentrations used are described in Table 1. All samples were tested with a 2-fold ACE2 dilutions, starting at 16 µg/mL. Data for samples at DFSO of 3, 12, and 30 days are shown. The MFI signals for RP1 IgM (orange) and RP2 IgG (blue) are shown in A (DFSO 3), C (DFSO 12), and E (DFSO 30). The ACE2 percent residual MFIs are shown in B (DFSO 3), D (DFSO 12), and F (DFSO 30). Signals representing a >30% decrease as compared to No ACE2 controls are highlighted light green. Please click here to view a larger version of this figure.
