1. Starting, stopping, and processing time course CFME reactions for HPLC-RID quantification.
- Thaw previously prepared E. coli lysates and prepare the rest of the reaction components on ice.
NOTE: The lysates reported here were derived from E. coli BL21DE3-Star grown in 2xYPTG (1.8 % glucose) media to mid-log phase.- Prepare an appropriate volume of filter-sterilized (0.20 µm pore filter) S30 buffer (1 M Tris-OAc adjusted to pH 8.2 with glacial acetic acid, 1.4 M Mg(OAc)2, and 6 M KOAc).
- Prepare an energy mix containing glucose, glutamate salts, ATP, Coenzyme A, NAD+, Bis-Tris buffer, and dipotassium phosphate in the S30 buffer. Final concentrations in the desired reaction volume used to prepare CFME reactions here were 100 mM glucose, 18 mM magnesium glutamate, 15 mM ammonium glutamate, 195 mM potassium glutamate, 1 mM ATP, 0.2 mM Coenzyme A, 1 mM NAD+, 150 mM Bis-Tris, and 10 mM dipotassium phosphate.
- Combine the components in 1.5 mL microcentrifuge tubes to prepare final reactions with 4.5 mg/mL of total lysate protein. Here, CFME reactions were prepared with final volumes of 50 µL in triplicate per timepoint. Incubate the reactions at 37 °C for their respective timeframes.
NOTE: Work fast and add lysate as the final component of the reaction mix to prevent premature metabolic reactions with glucose and glutamate salts. Minimal glucose consumption can occur depending on how long reaction mixes are incubated on ice. - Terminate the reactions and process the samples for HPLC-RID analysis.
- To terminate the triplicate reactions at their appropriate time points, immediately add an equal volume of 5% trichloroacetic acid to each sample's final reaction volume (i.e., 50 µL of 5% trichloroacetic acid to a 50 µL reaction). Dilute each sample with sterile water at 2x the reaction volume (i.e., 100 µL).
- To recapitulate time zero, mix the same volume of 5% trichloroacetic acid as the total final reaction volume (i.e., 50 µL) with the lysate prior to adding the rest of the reaction components. This acidification step precipitates lysate enzymes before they significantly metabolize glucose.
- Vortex the samples and centrifuge on a benchtop microcentrifuge at 11,600 x g for 5 min and transfer supernatants containing the organic analytes to clean tubes. Store the samples at -20 °C if HPLC analyses are to be conducted on a different day. Ensure to thaw the stored samples on ice before proceeding to the next step.
- Filter each supernatant with a 0.22 µm pore filter. As an alternative to syringes, use centrifuge tube filters and centrifuge the supernatants at 16,300 x g for 1 min.
- Transfer each filtrate to a clean HPLC glass vial. Load vials onto the HPLC autosampler tray.
- Prepare samples for standard curve generation.
- Prepare a stock solution of all target analytes dissolved in S30 buffer at equimolar amounts above the starting concentration of glucose in the CFME reactions. Here, a stock solution consisting of 150 µM glucose, succinate, lactate, formate, acetate, and ethanol, was prepared. Perform 1:1 (v/v) serial dilutions from the stock solution to obtain triplicate 50 µL solutions with final concentrations ranging from 0 µM to the stock concentration (i.e., 150 µM).
- Dilute each solution with 50 µL of 5% trichloroacetic acid and 100 µL of sterile water. Repeat steps 1.3.4-1.3.5.
NOTE: Run solutions for standard curve generation with each batch of samples to ensure accurate quantification of metabolite concentrations.
2. Preparing the HPLC system for metabolite detection.
- Under a fume hood, prepare a sterilized 5 mM sulfuric acid solution from deionized and filter-sterilized water. Add ~550 µL of a 98% HPLC grade sulfuric acid solution to 2 L of water to prepare 5 mM sulfuric acid.
CAUTION: Sulfuric acid is a hazardous chemical, and working under a fume hood with proper lab PPE prevents inhalation, skin contact, and eye contact. Concentrated sulfuric acid reacts vigorously with water and should be added directly to water, not the other way around. Store in a cool, dry area away from direct sunlight and follow proper waste disposal measures set by the laboratory. - Keep the 2 L bottle of 5 mM sulfuric acid incubated in a water bath next to the HPLC instrument. Set the water bath to 35 °C. Place tubing with a solvent filter in the solvent bottle and attach the other end to a degasser module in line with the pump module.
NOTE: Purging the system with a freshly prepared solvent prior to installing the column is good instrument handling practice. - Equip the HPLC instrument with the HPLC column in line with the RID module. Place the column in the 35 °C water bath if a column thermostat is not available.
- Prepare the RID module for analysis at 35 °C in Chromatography Data System (CDS) software installed on the system computer.
- In the View menu, select the Method and run Control View. Right-click on the Pump Module > Method. Set the flow rate to 0.55 mL·min-1 and select the On button to initiate the pump.
NOTE: If the column was in storage prior to being equipped on the HPLC, ramp up the flow rate to 0.55 mL·min-1 after equilibrating the column following the manufacturer's instructions. - Right-click on the panel corresponding to the RID Module > Method. Set the temperature of the RI detector module to 35 °C and select On to start warming up the RI detector module.
- Right-click on the panel RID Module > Control. Select On for the Purge Reference Cell for at least 15 min when using a fresh solvent or 1 h if different solvents flowed through the RI detector prior to this setup. Click on the On button.
NOTE: Keep the pump and RI detector On to achieve a stable baseline on the online plot. This is affected by temperature fluctuations in the laboratory and can take up to 4 h or longer. Keep the system On overnight prior to sample loading.
- In the View menu, select the Method and run Control View. Right-click on the Pump Module > Method. Set the flow rate to 0.55 mL·min-1 and select the On button to initiate the pump.
3. Creating a method for the isocratic HPLC separation of organic fermentation products in the CDS.
- From the menu bar, select Method > New Method. Select Method > Save Method as [MethodName].M. Select Method > Edit Entire Method > Instrument/Acquisition
- Within the Binary Pump tab, set the flow to 0.55 mL·min-1. Under Solvents, select the letter corresponding to the solvent input on the pump module and set it to 100% for isocratic elution. Set Pressure Limits to 0 and 400 bar and input 30 min as the Stoptime.
- Within the Sampler tab, set the Injection Volume to 50 µL. Select the As Pump/No Limit option under Stoptime. Set the Advanced Auxillary Settings for Draw Speed, Eject Speed, and Draw Position to 200 µL·min-1, 200 µL·min-1, and -0.5 mm.
- Within the RID tab, set Optical Unit Temperature to 35 °C. Under Signal, select Acquire for Signal and >0.2 min for Peakwidth. Select the As Pump/Injector option for Stoptime.
- Under Advanced within the RID tab, set Analog Output to 5% Zero Offset and 500,000 nRIU for Attenuation. Select the Positive option for Signal Polarity and the On option for Automatic Zero Before Analysis.
- Save the method by selecting Method > Save Method. Load the method by selecting Method > Load Method > [MethodName].M.
4. Creating a sequence table for autosampling and start the HPLC-RID system for data acquisition.
- From the menu bar, select Sequence > New Sequence Template. Select Sequence > Save Sequence Template as [SequenceTemplateName].S.
- Select Sequence > Sequence Table. Append 'n' rows corresponding to 'n' vials, then input vial positions and sample names under Vial and Sample Name, respectively, according to their arrangement on the autosampler tray. Select the Method generated in step 3 from the Method Name dropdown menu and input 50 µL as Inj/Vial (Injection per Vial) for each row.
- Click Apply and save the sequence template by selecting Sequence Template > Save Sequence Template. Ensure the sequence template is loaded by selecting Sequence > Load Sequence Template > [SequenceTemplateName].S.
- After achieving a stable baseline on the online plot, right-click the panel RID Module > Control > Off Recycling Valve to direct the solvent flow through the RID detector to waste. To start data acquisition, select Sequence from the menu bar, Sequence > Run.
5. Extracting and analyzing data post-run.
- Select Data Analysis view from the View menu. Locate the Sequence Filename from the File List on the left-hand side of the screen. On the center panel on the screen, go to the Signal View Selection > RID Signal to view the sample chromatograms.
- Select a row corresponding to a high concentration standard sample from the top panel on the screen. Take note of the retention times for the target analyte peaks on the displayed chromatogram. Peaks corresponding to the target analytes will be arranged along the retention time axis as glucose, succinate, lactate, formate, acetate, and ethanol (Supplemental Figure 1).
NOTE: The first large peak on the chromatogram corresponds to trichloroacetic acid. Its RI units should be consistent across all standard curve samples. Validate the retention time of each target analyte by running each compound as a separate sample. - Extract peak areas for each target analyte from chromatograms of the standards and the reaction samples.
- Discern whether the peaks-of-interest are well integrated by the software. Draw the red line as the base of each peak to obtain an accurately integrated area under the curve. If automatic integration fails (i.e., red line is askew), select the Manual Integration button from the Integration Tool Set and manually draw a peak base to integrate the peak area.
NOTE: If manual integration must be performed for a target analyte in one sample, keep consistent and manually integrate the same analyte across all samples. - Select the Cursor tool from the Common Tool Set to click on properly integrated peaks. The peak area and the corresponding retention time of the selected peak will be highlighted as a table row on the bottom panel of the screen.
- To export peak areas, select File > Export > Integration Results.
- Discern whether the peaks-of-interest are well integrated by the software. Draw the red line as the base of each peak to obtain an accurately integrated area under the curve. If automatic integration fails (i.e., red line is askew), select the Manual Integration button from the Integration Tool Set and manually draw a peak base to integrate the peak area.
- Quantify the target analyte concentrations using standard curves.
- Plot peak area values vs. known concentrations of samples in a spreadsheet. Right-click on the plotted data, Add Trendline > Format Trendline > Display Equation on Chart.
- In a separate spreadsheet, use the equations of standard curve trendlines to convert peak area values to concentrations for every analyte from each sample. Calculate the average peak areas and standard error values across triplicates for data visualization.
6. Starting, stopping, and processing time course isotope tracing CFME reactions for LC-MS/MS quantification.
- Set up triplicate reactions per time point (except time zero) on ice as described in 1.1-1.2. However, instead of glucose, use a final concentration of 100 mM 13C6-glucose in the reactions. Incubate the reactions at 37 °C for 1 h, 2 h, and 3 h.
- To terminate, flash freeze the reactions in liquid nitrogen and store them at -80 °C. Skip this storage step for same-day analysis.
NOTE: Trichloroacetic acid was not used to stop reactions due to interference from the acid when detecting some central carbon metabolites via LC-MS/MS. Instead, extraction solvent containing formic acid (step 6.3) was used to precipitate metabolic proteins since formic acid's mass is below the detection limit of the reported MS/MS method. - Prepare 50 mL of the extraction solvent. Combine and vortex 20 mL of acetonitrile, 20 mL of methanol, and 10 mL of water (all LC-MS grade) in a 50 mL centrifuge tube along with 0.199 mL of formic acid to make a 0.1 M solution. Chill the solvent to 4 °C during extraction and store the solvent at -20 °C when not in use.
- Processing samples for LC-MS/MS analysis
- On the day of analysis, pipette an equivalent volume of the extraction solvent (i.e., 50 µL) to each sample. If the samples were frozen, add the extraction solvent before the samples completely thaw out to prevent the reactivation of glucose metabolism. Perform all sample processing steps on ice.
- To recapitulate time zero, pipette the final volume of extraction solvent (i.e., 50 µL) to an appropriate volume of lysate for the desired final concentration in the reaction (i.e., of 4.5 mg/mL in 50 µL reaction volume). Add the rest of the reaction components as in step 1.2. This acidification step precipitates lysate enzymes before they significantly metabolize glucose.
- Incubate the samples in extraction solvent on ice for 30 min with gentle shaking, then centrifuge the samples at 21,000 x g for 15 min at 4 °C to separate the supernatant from the precipitated protein. Transfer 50 µL of the supernatant to autosampler vials and load the vials onto the tray within the 4 °C autosampler. Store the rest of the supernatant at -20 °C for future analyses.
7. Setting up the LC system for LC-MS/MS analysis.
- Prepare 1 L of Solvent A by completely dissolving 77.08 mg of ammonium acetate in 950 mL of water and 50 mL of isopropanol. Prepare 1 L of Solvent B with 650 mL of acetonitrile, 300 mL of water, and 50 mL of isopropanol along with 77.08 mg of ammonium acetate. Ensure that all solvents are LC-MS grade.
- Connect the solvent bottles containing Solvents A and B to the pump module. Purge the system at a high flow rate to remove/limit any air contamination that may have occurred during the equipment of the solvents to the LC system.
- Equip the system with a C18 reversed-phased column (30 cm column length, 75 µm inner diameter, and 5 µm particle diameter). Condition the column to the LC-MS system by flowing 100% solvent B and slowly flowing up Solvent A to 100%.
NOTE: Column tips were prepared in-house using a micropipette puller and packed with pressure cells and helium.
8. Creating a method on LC-MS/MS data acquisition and interpretation software for the LC system linked to Fourier Transform and Ion Trap Mass Spectrometers.
- Open the Tune Plus software to edit a tune file for the MS method.
- From the File on the menu bar, open a preinstalled negative mode tune file.
- Select ScanMode on the menu bar, then select Define Scan Window. Set the microscan time setting for MSn to 1 for both Ion Trap and FT.
- Go to settings for Nano-ESI Source and set Spray Voltage to 4 kV. Modulate this until an acceptable electrospray is generated; typically, acceptable electrospray can be achieved within the range of 2-5 kV.
- Save the tune file.
- Generate a new LC method using the Setup Wizard of the instrument's data acquisition and interpretation software. Open Roadmap > Sequence Setup > Wizard. Since these methods do not require the usage of a column heater, skip the Temp Control step.
- Under Flow Gradient Pump Options, select Multistep. In the next window, insert 7 lines and set the flow rate for each row to 0.1 mL·min-1. Input the following parameters for each row: from 0-3 min, deliver 100% solvent A; from 3-9 min, introduce a gradient from 100% solvent A to 20% solvent B; from 9-19 min, introduce a new gradient from 20% solvent B to 100% solvent B; from 19-27 min, hold at 100% solvent B; from 27-28 min, set the gradient back to 100% solvent A; from 28-44 min, rinse and recondition of the column for subsequent runs by holding at 100% solvent A. Include a final step to lower the flow rate to 0.03 mL·min-1 upon completion of the run to conserve the solvent when the LC is not in use.
- Apply Default settings for Sampler Options > Pump Pressure as the Acquisition Option and use Default Acquisition Time and use Default Pump Pressure options.
- Create an MS/MS method by selecting the Orbitrap Velos Pro MS icon from the sidebar on the Instrument Setup window.
- Click on New Method > Data Dependent MS/MS. Set Acquire Time to the length of the LC run (i.e., 44 min), Segment to 1, and Scan Events to 11. For Tune File, select the edited file from step 8.1.
NOTE: The first event is an MS1 precursor scan using the Fourier Transform MS (FTMS). The succeeding 10 events will be MS2 scans selecting the 10 most intense and unique ions in each precursor scan for MS2 fragmentation. - For Event 1, under Scan Description set Analyzer to FTMS and Polarity to Negative. Under MSn Settings, use a Resolution of 30,000 and a Normalized Collision Energy of 35 V. Set Scan Ranges to 50 m/z for first mass and 1800 m/z for last mass to capture small molecules.
- For events 2 through 11, under Scan Description set Analyzer to Ion Trap. Select Dependent Scan and click on Settings > Global > Dynamic Exclusion and select Enable; set a 30 s repeat duration and 120 s exclusion duration to eliminate repeat scans in proximity.
- Go to Scan Event settings and set Mass Determined from Scan Event to 1 for all MS2 events (2 through 11). To scan for the top 10 most intense ions, set each MS2 scan event to detect an nth Most Intense Ion from 1st to 10th. Therefore, set Event 2 to detect 1 as the nth Most Intense Ion, event 3 to detect 2, and so on.
- Close the setup window and go to File > Save As [Method_Name].meth.
NOTE: For general use, maintenance, and calibration of the LC instrument and mass spectrometer, refer to the operating instructions and manuals supplied by the manufacturer.
- Click on New Method > Data Dependent MS/MS. Set Acquire Time to the length of the LC run (i.e., 44 min), Segment to 1, and Scan Events to 11. For Tune File, select the edited file from step 8.1.
9. Setting up a run sequence and starting the LC-MS/MS run.
- Set up a Run Sequence using the LC-MS/MS system's data acquisition and interpretation software. Within Roadmap > Sequence Setup, right-click on the table to insert as many rows as samples. For each row, set the Inj Vol to 5 µL and the Position to the vial's respective position on the autosampler tray. Input file names as sample names and set the desired file path for run results.
NOTE: Blank vials containing Solvent A can be run at the start of the sequence and between each set of triplicate samples (each set of time points) to rinse the column. - To start the run, highlight all File Names in the Sequence. From the menu bar, select Actions > Run Sequence > OK.
10. Consolidating files and searching for tentative annotations on MZmine 2.53.
- Open MZmine and import the '.raw' output files from step 9.1. From the menu bar, select Raw Data Methods > Raw Data Import. Select files corresponding to the samples.
- Construct a list of peaks distinguishing between MS1 and MS2 scans. From the menu bar, Raw Data Methods > Feature Detection > MS/MS Peaklist Builder. Relevant settings include m/z Window set to 0.01 and Time Window set to the length of the run. Under set filters, select Negative as Polarity and Centroided as the Spectrum Type.
- From the menu bar, go to Feature List Methods > Feature Detection > Peak Extender. Set m/z Tolerance to 0.005 m/z or 10 ppm and Minimum Height to 1E3. This step will create fully fleshed-out peaks.
- Remove duplicate peaks. Go back to Feature List Methods > Filtering > Duplicate Peak Filter. Relevant settings include m/z Tolerance set to 0.005 m/z or 10 ppm and the RT Tolerance set to 5 min.
- To align peaks within similar data files (i.e., those of triplicate reactions), go back to Feature List Methods > Normalization > Retention Time Calibration. Be sure to process triplicate samples together and leave out blanks. Relevant settings include m/z Tolerance set to 0.005 m/z or 10 ppm, RT Tolerance set to 3 min absolute (min), and Minimum Standard Intensity set to 1E3.
- Align peaks from all files by m/z and retention time from Feature List Methods > Alignment > RANSAC Aligner. Set m/z Tolerance to 0.005 m/z or 10 ppm, RT Tolerance and RT Tolerance After Correction to 44 and 39 min, respectively, RANSAC Iterations to 0, Minimum Number of Points to 10%, and Threshold Value to 1. Tick the option Require Same Charge State.
- Correct for any data points that may have been lost in prior steps in Feature List Methods > Gap Filling > Peak Finder. Relevant settings include Intensity Tolerance set to 50%, m/z Tolerance set to 0.005 m/z or 10 ppm, and RT Tolerance set to 3 min. Enable RT correction.
11. Calculating negative mode masses of 13C-labeled glucose-derived metabolites and searching for the m/z features of these analytes in filtered data.
- Calculate the masses of 13C-labeled metabolites from glucose metabolism for the targeted search.
- Calculate the monoisotopic mass of each target compound from the number of atoms in the compound's molecular formula and the monoisotopic masses of each element20.
- Calculate the compound's negative mode mass [M-H]- by subtracting the mass of 1 proton (1.007276 Da) from the monoisotopic mass. This is the mass detected by negative mode MS detection after molecules are stripped of a hydrogen ion during ionization.
- From the negative mode mass, calculate the mass of the 13C-incorporating metabolite. Here, the masses of isotopologues that maximally incorporate glucose-derived 13C-labels were calculated.
- Use calculated masses of 13C-labeled metabolites to search and annotate m/z features from MZmine results. For each possible hit, calculate the mass error (ppm) using the following equation:
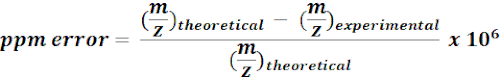
NOTE: Experimental m/z values with <15 ppm mass error were considered as putative annotations in the current analysis. - Manually check spectra of putative annotations on a quality browser to confirm annotations.
- Open Roadmap > Qual Browser. From the Tool Bar, Open Raw File to import raw MS data of each sample.
- Draw a line under the desired range of retention times (i.e., corresponding to the putative annotation) on the total ion chromatogram (top panel) to view a mass spectrum (bottom panel). Right-click on the spectrum and input a range of masses that encompass the target analyte's m/z. Check whether the putative annotations have distinct peak signals that are appreciably above the noise (Supplemental Figure 2).
- Calculate the average peak areas and the standard errors of positive annotations across biological replicates for each time point. Visualize the data (i.e., on a bar graph) to observe trends in glucose metabolism.
To quantify the lysate-based cell-free synthesis of common fermentation products from glucose, lysates derived from strains grown in 2xYPTG media were fed 100 µM glucose as a primary carbon source8. Reactions were stopped over a 24 h time course by protein acidification. Filtered supernatants containing pyruvate, succinate, lactate, formate, acetate, and ethanol produced from glucose catabolism were loaded onto the autosampler module of a HPLC system equipped with a RID module. Vials with filtered mixtures of fermentative end-products and glucose at 1.17 µM, 2.34 µM, 4.69 µM, 9.38 µM, 18.75 µM, 37.50 µM, 75 µM, and 150 µM concentrations in S30 buffer were loaded onto the instrument as standards. Analytes were eluted isocratically from an HPLC column to the RID. Peaks for glucose, succinate, lactate, formate, acetate, and ethanol within the 1 to 150 µM range could be resolved by RID. Peak areas for glucose were derived by manual integration from the RID data for time course and standard curve samples. Extracted peak areas for succinate, lactate, formate, acetate, and ethanol were taken from automatically integrated signals. All standard curves (peak area vs. known concentration) had R2 values >0.99 and were linear throughout the range of concentrations used here.
Molar concentrations for all target analytes were calculated from their respective standard curves. Glucose was consumed within the first 3 h of the reaction and mainly fermented to lactate (Figure 2A,B). Ethanol accumulation also significantly occurred within the first 3 h of the reaction and stopped thereafter (Figure 2C). The observation of significant lactate and ethanol production with significant glucose consumption after 3 h was not unprecedented since lactate and ethanol production pathways allow the regeneration of 1 net mol NAD+ from glycolytic NADH required for continued glucose consumption through glycolysis (Figure 1). Lactate and ethanol can thus be considered as the major fermentation end-products in lysate-based cell-free glucose metabolism. Acetate was initially present in the reactions as a component of the S30 buffer and unexpectedly only accumulated due to metabolism after 6 h when glucose consumption had slowed down (Figure 2D). This result suggests that acetate fermentation does not necessarily enable rapid glycolytic flux in earlier time points. Meanwhile, formate and succinate were synthesized as minor fermentation products (Figure 2E,F). Altogether, the method enabled the absolute quantification of sugar substrate depletion and fermentative product formation in E. coli S30 lysates.
MS detection to profile lysate glucose metabolism specifically was applied here. Lysates derived from strains grown in 2xYPTG media were fed 13C6-glucose as a carbon source. CFME reactions were run in triplicate for 0 h, 1 h, 2 h, and 3 h. Samples from each timepoint were loaded on an LC system equipped with a reversed-phase column and coupled to Fourier transform and ion trap mass spectrometers. Negative ion mode spectra were obtained and processed to analyze organic acids, sugar phosphates, and amino acids. Calculated theoretical masses of 13C-labeled species belonging to central carbon metabolism were searched to identify specifically glucose-derived compounds. Based on the utilized source strain cultivation conditions and previous reports of active pathways in E. coli CFME, it is assumed here that the lysate proteome comprises a metabolic network that feeds glucose into glycolytic fermentation, the pentose phosphate pathway, and possibly amino acid anabolism5,6,7,8,14 (Figure 1). Therefore, the search was narrowed down to members of these pathways, of which 16 metabolites incorporating glucose-derived 13C labels were unambiguously annotated (Supplemental Table 1).
13C6-glucose was observably consumed through glycolysis, as evidenced by the fluctuations in glycolytic intermediate abundances (Figure 3A–E). Consistent with the HPLC-RID data, glucose accumulated to 13C3-lactate and was also fermented to 13C3-succinate within the first 3 h of the reaction (Figure 4A,B). The formation of 13C3-succinate isotopologue supports the proposed model of lysate glucose metabolism (Figure 1), where succinate is likely to be generated by the carboxylation of 3-carbon phosphoenolpyruvate (PEP) molecule and not from the entry of a 2-carbon acetyl-CoA molecule to the TCA cycle. Activation of the TCA cycle has been assumed in previous CFME studies, but other 13C-labeled intermediates of TCA were not detected here8,19,21. 13C3-aspartate synthesis however occurred within the first h and was consumed, reinforcing the idea that PEP is directly converted to oxaloacetate (Figure 1, Figure 6C). The data are reflective of a lysate proteome from source strains harvested during fermentative growth on glucose-rich media (2xYPTG). This would further imply that the rest of the TCA enzymes not participating in succinate production form an oxidative TCA branch (Figure 1). None of the metabolites in this pathway, however, were detected, possibly because high concentrations of glutamate added to the CFME reaction as a salt counterion prevent the progression of this branch.
The HPLC-RID data is additionally complemented by the lack of 13C2-acetate detection within the 3 h reaction timeframe suggesting no build-up of acetate from glucose up to 3 h (Figure 2B). However, the direct precursor of acetate, acetyl-phosphate (acetyl-P), accumulated, suggesting that the Pta arm of the Pta-AckA pathway for acetate synthesis from acetyl-CoA is active (Figure 4C,D). The AckA catalyzed dephosphorylation of 13C2-acetyl-P to 13C2-acetate likely does not occur within this timeframe due to acetate being a major component of the S30 buffer used in the reactions (Figure 1, Figure 2B).
The incorporation of 13C6-glucose-derived carbons to sugar phosphates 6-phosphogluconolactone (6PGL), 6-phosphogluconate (6PG), ribulose-5-phosphate (Ru5P), and sedoheptulose-7-phosphate (S7P) was also observed (Figure 5). These results confirm the participation of the pentose phosphate pathway in lysate glucose metabolism and likely feeds 13C9-tyrosine synthesis, which has been suggested before by a proteomic study, while also providing a precursor for 13C5-histidine production (Figure 6A,B)7. Labeled phenylalanine and tryptophan were not observed here, and neither were most of the essential amino acids. However, this is not entirely surprising since amino acid anabolism is likely to be enriched in lysates derived from cells grown in nutrient-starved conditions or at the stationary phase7,22. Moreover, the data thus far suggests that intermediates of glycolysis and fermentation are funneled towards cofactor regenerating end reactions, which must preclude the synthesis of many amino acids derived from glyceraldehyde-3-phosphate, pyruvate, and acetyl-CoA (i.e., glycine, cysteine, serine, alanine, valine, leucine, and lysine) (Figure 1). As mentioned, 13C3-aspartate was produced within the first hour, whereas aspartate derived 13C-incorporating amino acids (i.e., threonine, isoleucine, methionine, and asparagine) were not observed possibly because glucose-derived aspartate participates in fermentation (Figure 1, Figure 6C). Lastly, flux towards labeled glutamate and amino acids derived from glutamate may have been impeded by high levels of glutamate in the reaction environment (Figure 1).
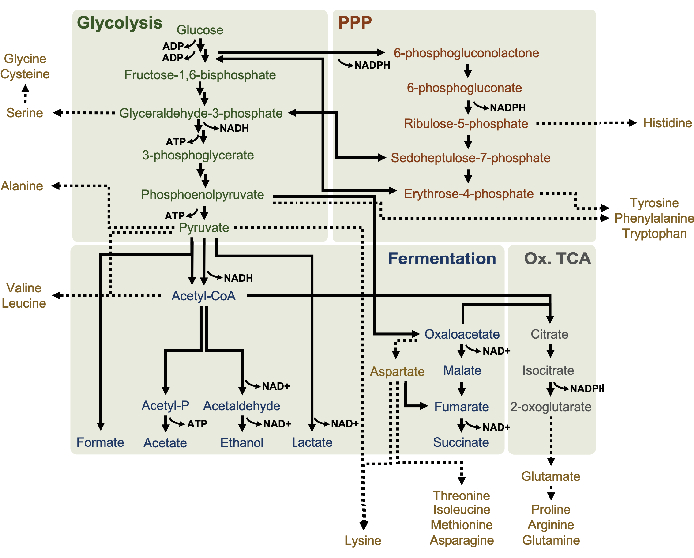
Figure 1: A putative metabolic model of lysates derived from E. coli BL21DE3-Star growing exponentially in high glucose concentrations. Intermediates and end-products of glycolysis (green), the pentose phosphate pathway (dark orange), and fermentative pathways (blue) from acetyl-CoA have been reported in lysate-based CFME. The presence of succinate fermentation implies the activation of the oxidative TCA branch (gray). Amino acid anabolism (gold) in lysates is not well-defined and is investigated here. Please click here to view a larger version of this figure.
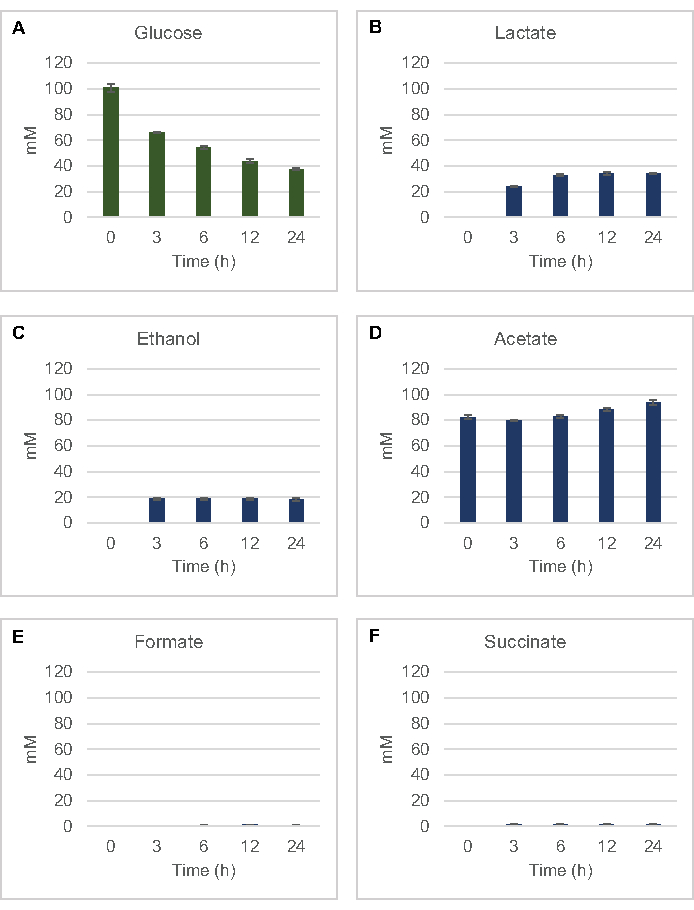
Figure 2: HPLC-RID data for glucose consumption and fermentative end-product synthesis in CFME reactions prepared with E. coli crude extracts. (A) Glucose consumption and (B) lactate, (C) ethanol, (D) acetate, (E) formate, and (F) succinate production in CFME reactions were monitored over 24 h. Average mM concentrations and error bars (SE) quantified with standard curves are presented (n = 3). Please click here to view a larger version of this figure.
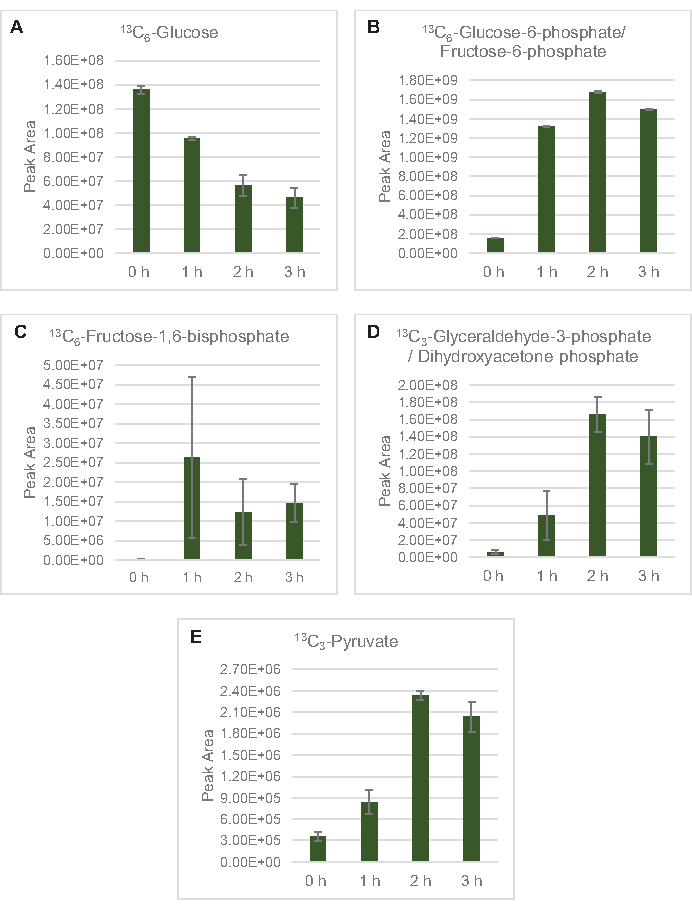
Figure 3: Time course trends of 13C6-glucose and 13C-labeled glycolytic intermediates in E. coli lysate CFME. Relative abundances of (A) 13C6-glucose, (B) 13C6-glucose-6-phosphate/fructose-6-phosphate, (C) 13C6-fructose-1,6-bisphosphate, (D) 13C3-glyceraldehyde-3-phosphate/dihydroxyacetone phosphate, and (E) 13C6-pyruvate in CFME reactions over 3 h. Raw peak areas extracted by mzMINE software were used to calculate averages and error bars (SE) for positive annotations (n = 3). Please click here to view a larger version of this figure.
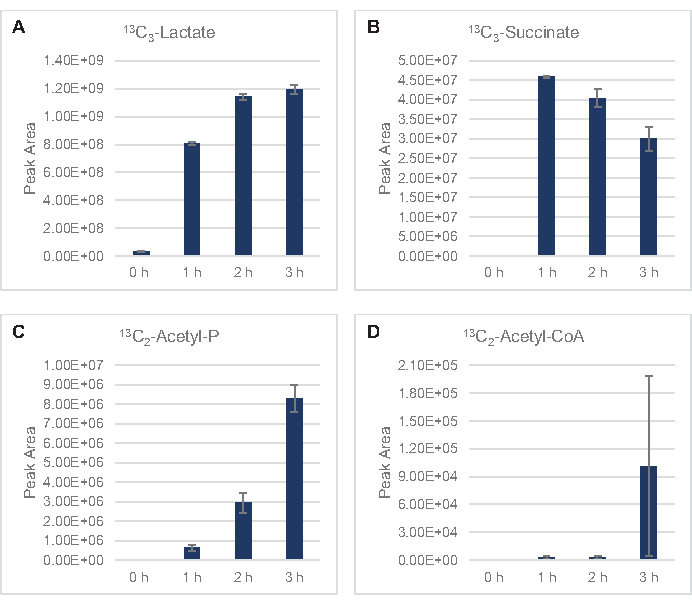
Figure 4: Time course trends of intermediates and end-products in 13C6-glucose fermentation in E. coli lysate CFME. Relative abundances of (A) 13C3-lactate, (B) 13C3-succinate, (C) 13C2-acetyl-phosphate, and (D) 13C2-acetyl-CoA in CFME reactions over 3 h. Raw peak areas extracted by mzMINE software were used to calculate averages and error bars (SE) for positive annotations (n = 3). Please click here to view a larger version of this figure.
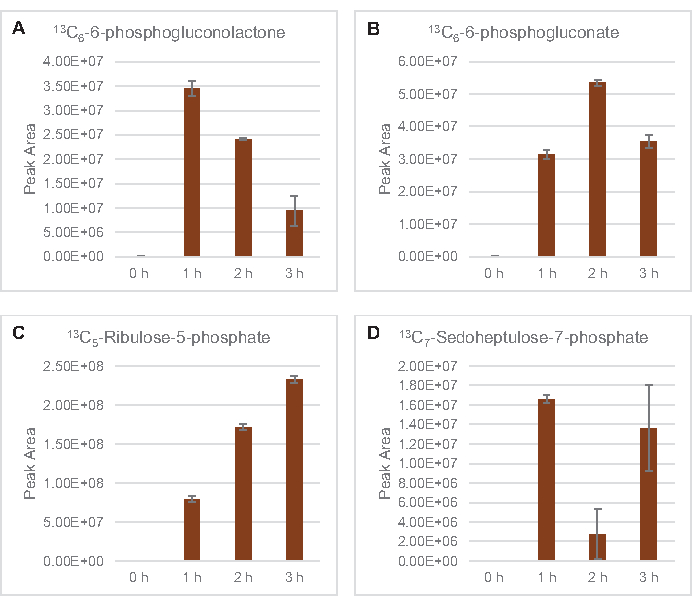
Figure 5: Time course trends of 13C6-glucose derived pentose phosphate pathway intermediates in E. coli lysate CFME. Relative abundances of (A) 13C6-6-phosphogluconolactone, (B) 13C6-6-phosphogluconate, (C) 13C5-ribulose-5-phosphate, and (D) 13C7-sedoheptulose-7-phosphate over 3 h. Raw peak areas extracted by mzMINE software were used to calculate averages and error bars (SE) for positive annotations (n = 3). Please click here to view a larger version of this figure.
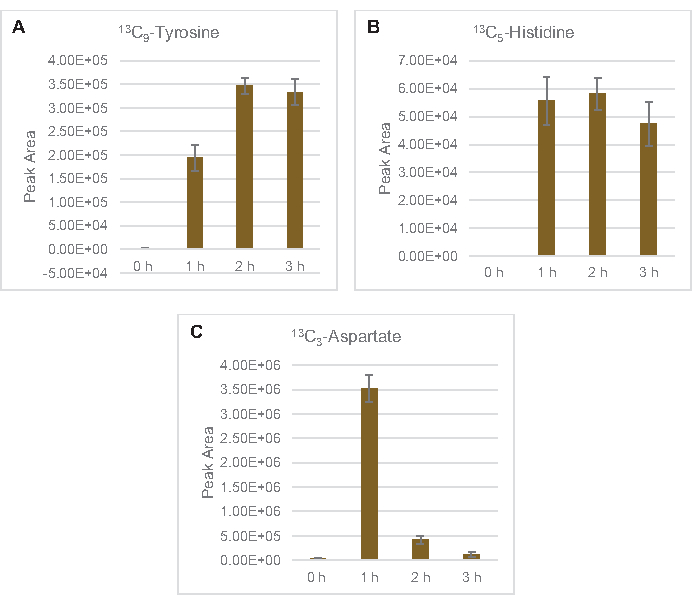
Figure 6: Time course trends of detected 13C6-glucose derived amino acids in E. coli lysate CFME. Relative abundances of (A) 13C9-tyrosine, (B) 13C5-histidine, and (C) 13C3-aspartate over 3 h. Raw peak areas extracted by mzMINE software were used to calculate averages and error bars (SE) for positive annotations (n = 3). Please click here to view a larger version of this figure.
Supplemental Figure 1: Representative HPLC-RID chromatogram showing peaks for major fermentative products in a CFME reaction incubated at 37 °C for 24 h. Glucose, succinate, lactate, formate, acetate, and ethanol peaks are sufficiently distinguishable by their retention times on an HPLC column during isocratic elution with 5 mM sulfuric acid solvent. Please click here to download this File.
Supplemental Figure 2: Representative mass spectra for 13C-labeled metabolites, specifically (A) lactate,(B) glucose, and (C) 6-phosphogluconate (6PG) in a CFME reaction incubated at 37 °C for 1 h. Please click here to download this File.
Supplemental Table 1: List of detected 13C-labeled metabolites, retention times (aligned across samples using MZmine), theoretical fully 13C-labeled negative mode m/z values, m/z values of detected features, and calculated mass errors. Please click here to download this Table.
