The present article represents a multipurpose device (MD) and complementing methodologies for culturing, freezing, thawing, histological staining, immunohistochemical staining, immunofluorescence labeling, coating, and processing of entire organoids or spheroids while still in a hydrogel in a single uniquely designed environment. The current study was designed to prepare HepG2 liver cancer spheroids in 35 hydrogel drops in 35 MDs. Experiments were conducted in triplicate to ensure accuracy. Additionally, lung organoids in MDs were immunofluorescent labeled as an example to demonstrate the results of the current methodologies for organoid studies.
Figure 1 shows a close look at the device containing a hydrogel dome. The niche's size and shape are designed to provide a protective environment for the hydrogel containing the organoids/spheroids and save the reagents used during various processes. In addition, the side part of the device that surrounds the niche can be used as the humidity chamber during immunostaining experiments. The medium to feed the sample may vary between 100-200 µL, depending on the height of the hydrogel drop. The current study focuses on the dome-based method since many hydrogels, commercial extracellular matrix components, and basement membrane extracts allow the user to prepare drops. However, it is also possible to fill the niche of the MD with the hydrogel and then seed the cells inside it to generate organoids/spheroids. That method might be preferred if the viscosity of the matrix does not allow the preparation of domes or if the experiment includes large volumes of organoids. The hydrogel type and the hydrogel/medium ratio for the preparation of drops may vary according to the cell type, the experimental design, and the medium. Figure 1 also represents the device's design that makes it possible to investigate the organoids/spheroids under brightfield, confocal, and scanning electron microscopes while organoids/spheroids are still in their native in vitro environment.
Figure 2 presents how to seed cells in a hydrogel and examine the development of spheroids or organoids. The design and dimensions of the multipurpose device have also been presented in that figure. Video 1 demonstrates the location of 3D growing spheroids in a hydrogel. Figure 3 represents live images of growing spheroids from day 3 to day 21. The time for the formation of spheroids and organoids may vary according to the nature of the sample. For example, spheroids from HepG2 cells and HEK cells formed within 3 days, while the formation of liver and biliary organoids lasted 2 weeks during the experiments.
Hematoxylin and Eosin-stained spheroids are seen in Figure 4. The image represents well-preserved and homogenously stained spheroids at different sizes and fusions of spheroids. Live cells in the center of the spheroids are noteworthy. The image also demonstrates the delicate connections between the cells from different spheroids. These fragile connections could not be visualized after traditional workflows since transferring, pipetting, or centrifuging would damage them. Figure 5 demonstrates immunostaining of spheroids with the antibody specific for Arginase, one of the most common markers for the diagnosis and prognosis of hepatocellular carcinoma42,43. The micrographs reveal differentiated and undifferentiated liver cancer cells in the same spheroids. This figure contains images with and without counterstaining with Hematoxylin. Researchers might choose to omit counterstaining with Hematoxylin in cases in which it would be hard to differentiate labeled areas in a 3D structure.
Figure 6 represents another straightforward protocol for whole-mount visualization of living organoids/spheroids in a hydrogel: live cell membrane and nucleus staining. The methodology allows the same labeling density in the periphery and the center, showing complete reagent penetration. Figure 7, Figure 8, and Figure 9 show representative images of spheroids labeled with one, two, or three antibodies, respectively. One to three primary antibodies are diluted in S-IF simultaneously. Similarly, one to three matching secondary antibodies suitable for the experiment are diluted simultaneously in S-IF. The immunolabeling protocol allows researchers to mark the 3D structures within 4-6 h without losing or damaging them. The background is transparent, and the technology used here does not require additional clearing, antigen retrieval, or blocking methods/solutions. The method also allows researchers to label the specimen with multiple antibodies in a single step. In other words, the user prepares one solution containing one to three primary antibodies and another solution matching with one to three secondary antibodies. The protocol described here eliminates sequential labeling steps with different antibodies in conventional methodologies.
Figure 10 represents scanning and transmission electron microscopic images of the spheroids. The first row demonstrates pictures of entire spheroids in the MD under a scanning electron microscope. The second row contains transmission electron microscopic images of spheroids after preparation of the resin blocks containing whole-mount spheroids in the MD, as well as images of sectioned and stained spheroids. Well-preserved cytoplasmic organelles and other ultrastructural features of the cells demonstrate the effectiveness of this straightforward protocol, which protects the 3D structure of the whole sample. The MD also allows the researchers to freeze and thaw the whole-mount samples in a hydrogel. Figure 11 demonstrates hydrogel domes and the spheroids at a higher magnification before and after freezing. The irregularity at the boundaries of the hydrogel dome is notable. However, the roundness of the cryopreserved spheroids is nearly stable after thawing compared to the spheroids before freezing. Live cell membrane and nucleus labeling methods are also applied 48 h after thawing to demonstrate how the freezing/thawing procedure affected the 3D architecture, the cell membrane, and the cell viability. The traditional freezing solution containing dimethyl sulfoxide and the current newly formulated solution, SF, demonstrate similar results. More than 75% of the 3D structures could survive in this protocol. However, further experiments are needed to reveal the long-term side effects of each formulation on organoids and spheroids.
Table 1 and Table 2 compare the traditional workflows with those described here based on step number, duration, and waste production (i.e., the total number of plastic gloves, pipette tips, serological pipettes, centrifuge tubes, microcentrifuge tubes, cell culture vessels, cryovials, etc. for each workflow).
Supplementary Figure 1 represents sequential steps that can be performed in a single MD schematically. Supplementary Figure 2 demonstrates the results of the experiments designed to compare the cell viability, toxicity, and proliferation rates of HepG2 cells in an MD or a traditional glass-bottomed dish. Supplementary Figure 3 shows the foam boxes that have been used for freezing the samples in MDs. Supplementary Figure 4 summarizes the steps to take out a resin block from an MD. Supplementary Figure 5 includes images of airway organoids that were immunofluorescent labeled and then visualized while they were still in MDs. Similarly, Video 2 demonstrates two immunofluorescent labeled airway organoids in an MD. Finally, Supplementary Figure 6 is a graph comparing the mean intensity of cell membrane labeling intensity in live spheroids before and after freezing using traditional workflow and the workflow described here. Image J software has been used for analysis.

Figure 1: The multipurpose cell culture device (MD). (A) The niche (N), the central part of the device, is designed to create a protective environment for the organoids/spheroids grown in a hydrogel drop (D) during sequential processes. The side (S), the surrounding part of the niche, can be used as a humidifier chamber during immunostaining experiments. (B) The transparent center in the lid allows the user to observe the organoids/spheroids when the device is closed. (C) The hydrogel dome containing organoids in the MD can be stained or embedded in a resin block for transmission electron microscopy. The lid also includes the niche (N) for hanging drop methodology. The size and the design of the device allow the user to examine the organoids/spheroids under (D) brightfield, (E) confocal, and (F) scanning electron microscopes. Please click here to view a larger version of this figure.
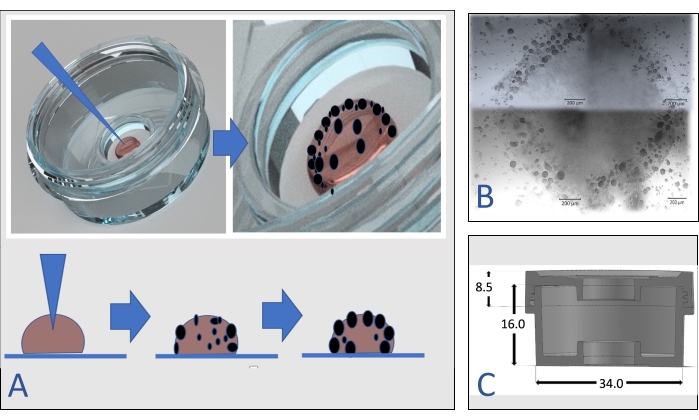
Figure 2: Seeding cells inside a hydrogel. (A) The cell pellet in the pipette is inserted into the top of the dome. After 3-5 days, spheroids become visible in the dome, especially at the periphery of the drop. (B) Four live images of growing spheroids from each quarter in a dome were captured and merged. Scale bar = 200 µm. (C)The design and dimensions (in centimeters) of the multipurpose device (MD). Please click here to view a larger version of this figure.
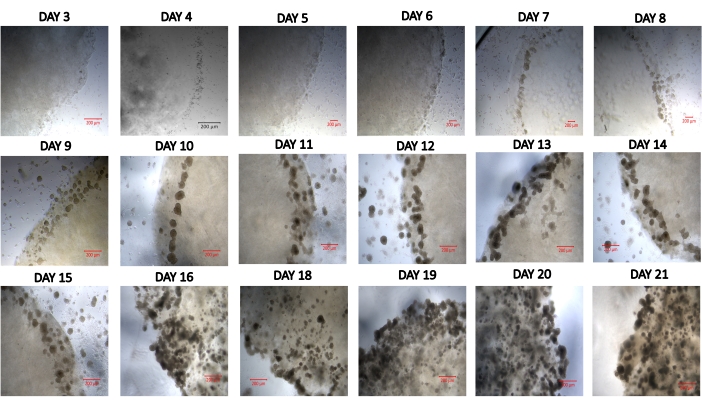
Figure 3: Developing spheroids in a hydrogel drop from day 3 to day 21. Scale bar = 200 µm. Please click here to view a larger version of this figure.
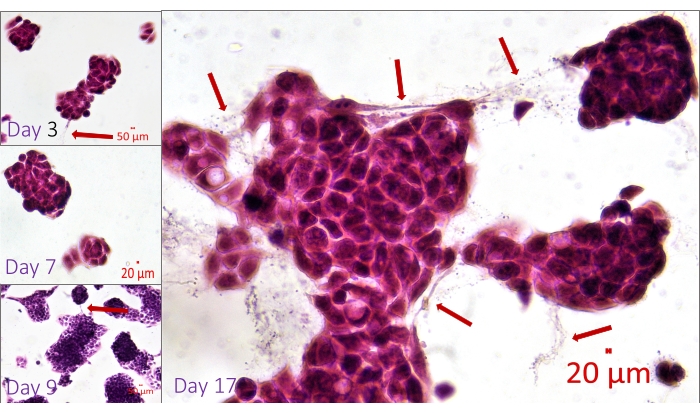
Figure 4: Hematoxylin and Eosin-stained whole-mount spheroids within the MD. Visualization of the connections between the cells located in the adjacent or fusing spheroids. The delicate processes between the cells (arrows) are visible since the whole-mount sample in the hydrogel is fixed, stained, and examined without damaging the 3D growing structures. Scale bar: day 3, day 9 = 50 µm; day 7, day 17 = 20 µm. Please click here to view a larger version of this figure.
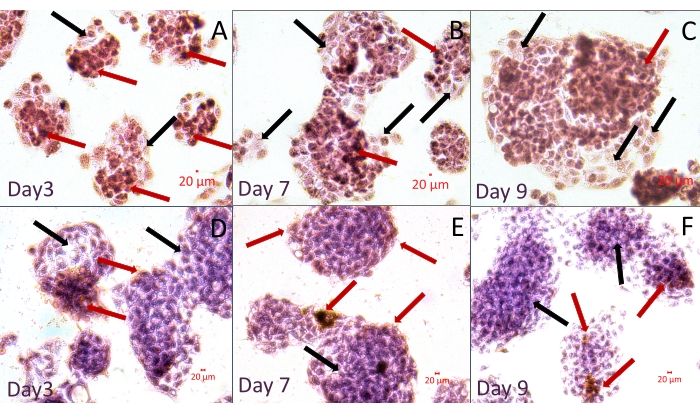
Figure 5: Immunohistochemical staining of whole-mount spheroids in the hydrogel within the MD. The samples were immunostained with an antibody specific to Arginase. Arginase positive (red arrows) and negative (black arrows) cells are seen in the spheroids. Images are from two experiments: (A–C) without and (D–F) with counterstaining with Hematoxylin. Scale bar = 20 µm. Please click here to view a larger version of this figure.
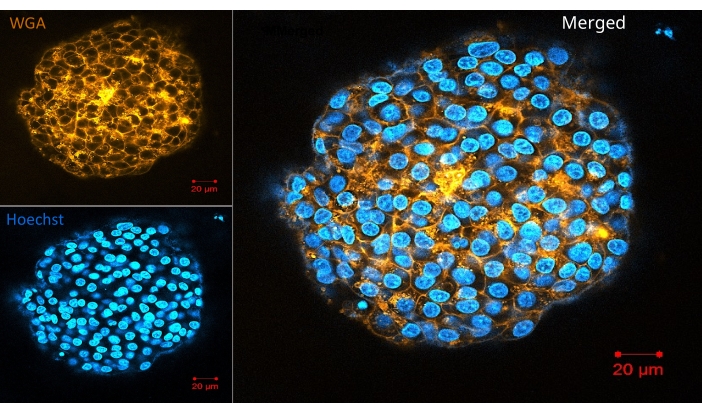
Figure 6: Confocal image of a live organoid in the hydrogel. The living organoids were labeled with the live cell membrane (WGA) and nucleus stains (Hoechst). Scale bar = 20 µm. Please click here to view a larger version of this figure.
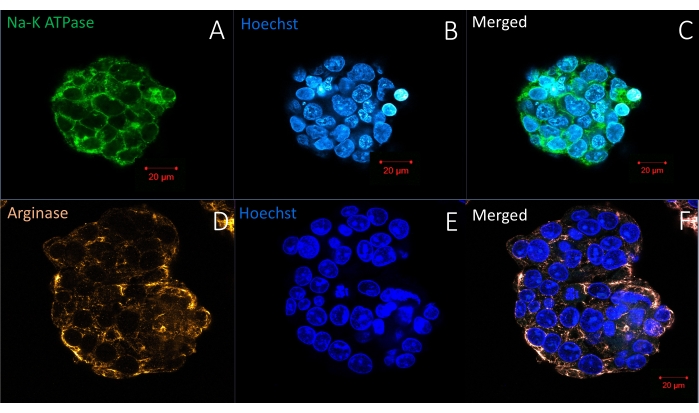
Figure 7: Immunofluorescence labeling of whole-mount spheroids in a hydrogel with one antibody and a nuclear stain (Hoechst). (A–C) The upper panel shows a spheroid labeled with an antibody specific for Na-K ATPase in the cell membrane. (D–F) The bottom panel demonstrates the location of another antibody specific for Arginase, a cytosolic protein specific for hepatocellular carcinoma, in liver cancer spheroids. Duration of staining protocol: 6 h. Scale bar = 20 µm. Please click here to view a larger version of this figure.
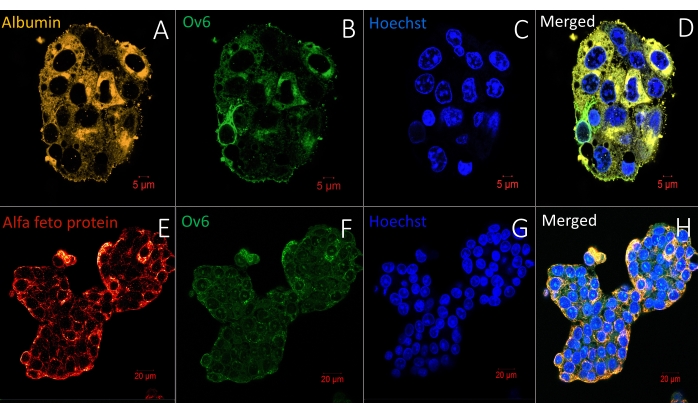
Figure 8: Immunofluorescence labeling of whole-mount spheroids in a hydrogel with two antibodies and a nuclear stain (Hoechst). (A–C) The upper panel shows a spheroid labeled with two antibodies specific for albumin and Ov6. Scale bar = 5 µm. (D–F) The bottom panel demonstrates the location of alpha feto protein and Ov6 in liver cancer spheroids. Scale bar = 20 µm. Duration of staining protocol: 6 h. Please click here to view a larger version of this figure.
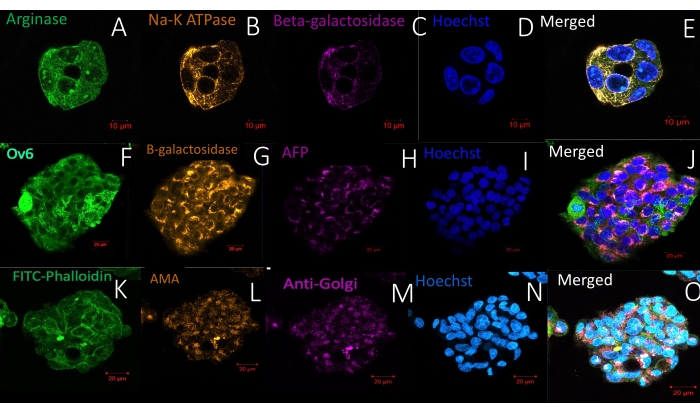
Figure 9: Immunofluorescence labeling of whole-mount spheroids in a hydrogel with three antibodies and a nuclear stain (Hoechst). (A–E) The spheroid in the upper panel is labeled with antibodies specific for Arginase, Na-K ATPase, and beta-galactosidase. Scale bar = 10 µm. (F–J) The middle panel demonstrates a spheroid labeled with Ov6, beta-galactosidase, and alpha-feto protein. Scale bar = 20 µm. (K–O) The spheroid in the lower panel has been labeled with FITC-phalloidin, anti-mitochondrial antibody, and anti-Golgi antibody. Scale bar = 20 µm. Note the high labeling specificity and reduced background. Duration of staining protocol: 6 h. Please click here to view a larger version of this figure.
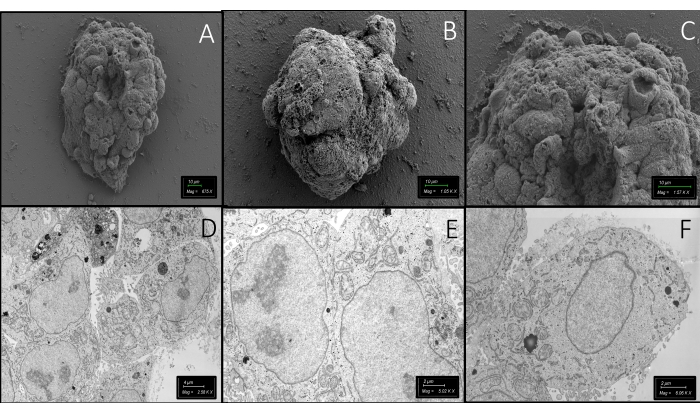
Figure 10: Electron microscopy images. (A–C) Scanning electron microscopic images of entire spheroids in the hydrogel in the MD. Scale bar = 10 µm. (D–F) Ultrastructural features of the cells in a spheroid have also been visualized under a transmission electron microscope. Scale bars: (D) = 4 µm; (E,F) = 2 µm. Please click here to view a larger version of this figure.
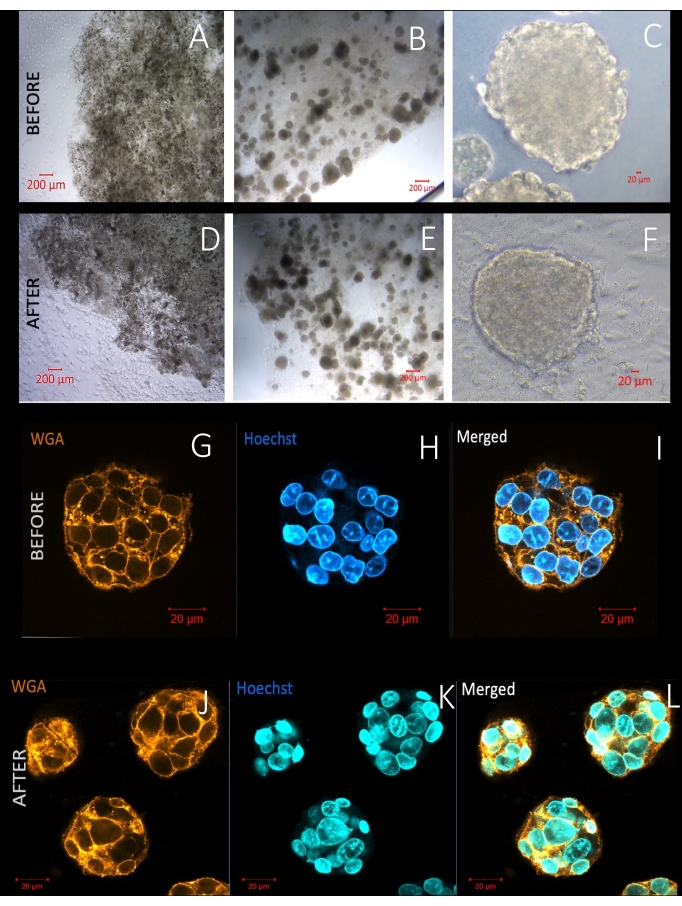
Figure 11: Live images of spheroids before and after freezing. (A–F) The irregularity of the borders of the hydrogel domes is evident after freezing. However, the size and roundness of spheroids remain similar. (F) Cell migration on the culture surface can be seen after thawing. (G–L) The live cell membrane (WGA) and nucleus staining (Hoechst) demonstrate similar cell viability and intact cell membranes before and after freezing the entire spheroids in a hydrogel in the MD. Scale basr: (A,B,D,E) = 200 µm; (C,F,G–L) = 20 µm. Please click here to view a larger version of this figure.
| Tradional workflow | Workflow with multipurpose device (MD) | |
| Steps | 22 | 6 |
| Time | 9 days | 4 days |
| Waste Products | 26 | 9 |
Table 1: Comparison of step number, time, and waste production between the traditional and new workflows during a short-term experiment.
| Traditional workflow | Workflow with Solution for immunofluorescence (S-IF) | |
| Steps | 25 | 7 |
| Time | 120 h | 6-8 h |
| Waste Products | 28 | 10 |
Table 2: Comparison of the conventional immunolabeling and the new immunolabeling protocol.
Video 1: Time series of images at different levels of a hydrogel containing spheroids under an inverted phase contrast microscope. Please click here to download this Video.
Video 2: 3D structure of two airway organoids labeled with antibodies for Cytokeratin 5 and DAPI. Please click here to download this Video.
Supplementary Figure 1: Overview of the experiment. This schematic shows the sequential experimental steps for growing and examining whole-mount organoids in a hydrogel. The technology described here makes it possible to grow, freeze, thaw, label, and examine the organoids under different microscopes. Please click here to download this File.
Supplementary Figure 2: Comparison of the cell viability, toxicity, and proliferation rates of HepG2 cells in a traditional glass-bottomed dish or an MD. HepG2 cells were grown on(A) a traditional glass-bottomed cell culture dish or (B) in an MD to compare cell viability and toxicity. (C) Trypan blue exclusion test was used to demonstrate the viability. Scale bar = 20 µm. The results were compared using (D–E) CCK8 cytotoxicity and (F) cell proliferation assays. Please click here to download this File.
Supplementary Figure 3: Arrangement of the two foam boxes. One foam box is placed inside the other during the slow freezing process of the entire organoids or spheroids in the MD. *denotes the two boxes. Please click here to download this File.
Supplementary Figure 4: Steps demonstrating how to remove the resin block from the MD after polymerization of the resin. (A) The resin block surrounding the spheroids or organoids is prepared inside the MD niche and then polymerized. (B–D) Then, with an appropriate thin rod, the resin block is pushed to remove it from the niche. (E) Next, the resin block, with the plastic below, is removed. (F–G) Finally, a pair of tweezers is used to separate them if the block is still attached to the plastic. (H) The block is ready for thin sectioning. The whole-mount organoids or spheroids in the resin block (*) are ready for sectioning for transmission electron microscopy. Please click here to download this File.
Supplementary Figure 5: Airway organoids that were immunofluorescent labeled and then visualized while they were still in MDs. (A–C) The upper panel shows circular airway organoids with a central lumen. Green represents airway progenitor cells expressing Cytokeratin 5 in the outermost ring of the organoid, and blue represents the nuclei. Scale bar = 50 µm. (D–F) The bottom panel shows the three-dimensional image of the two side-by-side organoids in the (D) DAPI and (E) Alexa 488 filters, as well as the (F) merged image. Scale bar = 100 µm. Please click here to download this File.
Supplementary Figure 6: Comparison of the labeling density on the live cell membranes of the cells. The graph compares the spheroids before and after freezing using the traditional and presently adopted workflow. Analysis was performed using the Image J image analysis software. Abbreviations: IntDen = Integrated Density (fluorescence intensity in the selected spheroids). CTCF = Corrected total cell fluorescence. Please click here to download this File.
