The IVT template is a 127 bp PCR product (Figure 1B). The full-length IVT product is a 98 nt RNA, which migrates similarly to a 70 bp double-stranded DNA fragment (Figure 1C).
After electroporation, the cells should be >90% viable, with a total cell count of >70% of the starting cell number. The resulting pool of mutant cells should have a diverse set of indels, starting near the Cas9 cleavage site. The analysis of the targeted genes by PCR and Sanger sequencing should show multiple nucleotides at each position downstream of the Cas9 cleavage site (Figure 2).
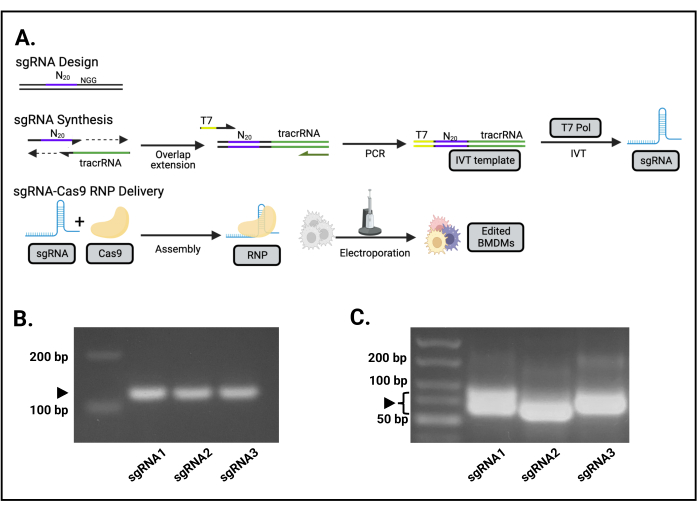
Figure 1: Overview of the sgRNA-Cas9 editing process. (A) Schematic for the guide design, sgRNA generation, and Cas9-sgRNA delivery. (B) The PCR product from IVT for Src sgRNAs 1, 2, and 3 resolved on a 2% agarose gel. The arrow indicates the correct 127 bp PCR products. (C) The RNA products following IVT for Src sgRNAs 1, 2, and 3 resolved on a 2% agarose gel. The brackets indicate the correct sgRNA products. The variable migration of the sgRNA is due to RNA secondary structure. This figure is reprinted from the first author's master's thesis16. Please click here to view a larger version of this figure.
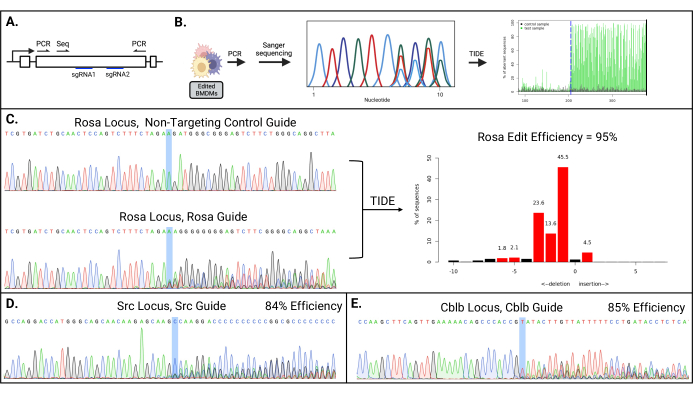
Figure 2: High editing efficiency achieved for multiple targeted genes. (A) Schematic of PCR and Sanger sequencing primers. (B) Schematic of the workflow for TIDE to assess the editing efficiency. (C) Representative Sanger sequencing chromatogram of the ROSA26 locus from BMDMs electroporated with a control non-targeting sgRNA-Cas9 RNP (top) and ROSA26-specific sgRNA (bottom); the Cas9 cleavage site is highlighted. The TIDE output (right) with the calculated editing efficiency and the percentage of sequences harboring the indicated number of indels. (D,E) Sanger sequencing chromatograms of the (D) Src and Cblb genes in the edited BMDMs. This figure is reprinted from the first author's master's thesis16. Please click here to view a larger version of this figure.
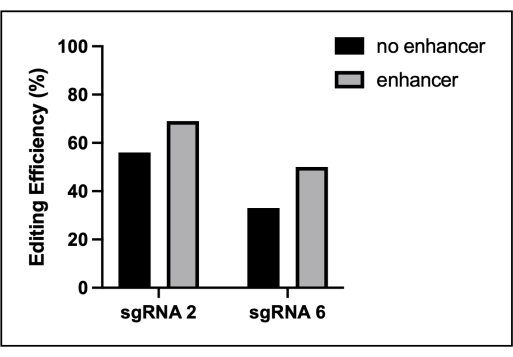
Figure 3: Moderate increase in editing efficiency due to the commercial electroporation enhancer. The BMDMs were electroporated with low-efficiency guides to evaluate the effect of a commercial editing enhancer. Prior to electroporation, 1 µL of the enhancer was added to the assembled RNPs to a final concentration of 4 µM. The editing efficiency of the indicated Src sgRNA was evaluated using TIDE. This figure is reprinted from the first author's master's thesis16. Please click here to view a larger version of this figure.
| Tool Name | URL | ||
| Synthego – CRISPR Design Tool | https://www.synthego.com/products/bioinformatics/crispr-design-tool | ||
| The Broad Institute – CRISPick | https://portals.broadinstitute.org/gppx/crispick/public | ||
| Tracking of Indels by Decomposition (TIDE) | http://shinyapps.datacurators.nl/tide/ | ||
Table 1: URLs of online tools.
| Primer Name | Sequence | ||
| Guide-Specific Primer | ggatcctaatacgactcactatag[N20]gttttagagctagaa | ||
| Guide-Specific Primer – Cbl-b Guide 3 | ggatcctaatacgactcactatagAAAATATCAAGTATATACGGgttttagagctagaa | ||
| Guide-Specific Primer – Cbl-b Guide 4 | ggatcctaatacgactcactatagGGTAAAATATCAAGTATATAgttttagagctagaa | ||
| Guide-Specific Primer – Rosa | ggatcctaatacgactcactatagCTCCAGTCTTTCTAGAAGATgttttagagctagaa | ||
| Guide-Specific Primer – Scramble | ggatcctaatacgactcactatagGCACTACCAGAGCTAACTCAgttttagagctagaa | ||
| Guide-Specific Primer – Src Guide 2 | ggatcctaatacgactcactatagTCACTAGACGGGAATCAGAGgttttagagctagaa | ||
| Guide-Specific Primer – Src Guide 5 | ggatcctaatacgactcactatagCAGCAACAAGAGCAAGCCCAgttttagagctagaa | ||
| Guide-Specific Primer – Src Guide 6 | ggatcctaatacgactcactatagAGCCCAAGGACGCCAGCCAGgttttagagctagaa | ||
| T7 Reverse Long Universal Primer | aaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac | ||
| Universal Forward Amplification Primer | ggatcctaatacgactcactatag | ||
| Universal Reverse Amplification Primer | aaaaaagcaccgactcgg | ||
Table 2: Oligonucleotides used in the PCR to generate the template for the IVT of the sgRNA. The 20 nucleotide target sequences for gene-specific primers are capitalized.
| BMDM Growth media. Store at 4 C. | |||
| DMEM | |||
| Fetal bovine serum | 0.1 | ||
| L-glutamine | 0.2 M | ||
| MCSF supernatant from 3T3-MCSF Cells** | 0.1 | ||
| Sodium pyruvate | 11 mg/mL | ||
| Lysis Buffer. Store at 4 C. | |||
| 2-mercaptoethanol (add immediately prior to use) | 0.01 | ||
| MgCl2 | 5 mM | ||
| Tris | 20 mM | ||
| Triton-X 100 | 0.005 | ||
| **3T3-MCSF cells are grown in DMEM+10% FCS. Supernatent with MCSF is harvested on the 5th day after reaching 100% confluence. As an alternative 10ng/ml recombinant MCSF can be used in lieu of conditioned media | |||
Table 3: Compositions of the media and buffers.
Supplementary File 1: Raw sequencing file for ROSA_ Mock Please click here to download this File.
Supplementary File 2: Raw sequencing file for ROSA_TargettingGuide Please click here to download this File.
Supplementary File 3: Raw sequencing file for ScrambleGuide Please click here to download this File.
Supplementary File 4: Raw sequencing file for SrcG5+SrcG6 Please click here to download this File.
Supplementary File 5: Raw sequencing file for CBLB_Mock Please click here to download this File.
Supplementary File 6: Raw sequencing file for CBLB_TargetingGuide Please click here to download this File.
Supplementary File 7: Raw sequencing file for Mock_Guide2Locus Please click here to download this File.
Supplementary File 8: Raw sequencing file for Mock_Guide6Locus Please click here to download this File.
Supplementary File 9: Raw sequencing file for SrcGuide2_Enhancer Please click here to download this File.
Supplementary File 10: Raw sequencing file for SrcGuide2_NoEnhance Please click here to download this File.
Supplementary File 11: Raw sequencing file for SrcGuide6_Enhancer Please click here to download this File.
Supplementary File 12: Raw sequencing file for SrcGuide6_NoEnhancer Please click here to download this File.
