1. Experimental preparation
NOTE: The details of all the reagents/equipment mentioned in this step are listed in the Table of Materials.
- Obtain recombinant human PD-1 (rhPD-1; polyhistidine-tagged). Reconstitute lyophilized rhPD1 with sterile-filtered phosphate-buffered saline (PBS), pH 7.4, before use.
- Obtain biotinylated recombinant human PD-L1 (rhPD-L1). Reconstitute lyophilized rhPD-L1 with sterile deionized water before use.
- Obtain streptavidin-conjugated R-phycoerythrin detection reagent (SAPE). Store all the SAPE solutions protected from light at refrigerator temperatures (i.e., 2-8 °C).
- Obtain fluorescently dyed magnetic microspheres (6.5 µm diameter, polystyrene with embedded magnetite) and the Bead Coupling Kit15 (if used, see the Table of Materials). Covalent coupling to the microspheres requires sulfo-NHS (sulfo-N-hydrosuccinimide) and EDC (N-[3-dimethylaminopropyl]-N′-ethylcarbodiimide hydrochloride).
NOTE: Magnetic bead sets are available with any of 500 unique fluorescent tags, which allows for identification and differentiation from different bead sets16. Beads are offered at stock concentrations of 2.5 × 106 beads/mL and 12.5 × 106 beads/mL. Store the beads protected from light at refrigerator temperatures (i.e., 2-8 °C). Do not freeze the bead suspensions. - Obtain positive and negative control antibodies and Brilliant Violet 421 (BV421)-labeled secondary detection antibodies. Store all the fluorescent-conjugated molecules protected from light.
- Perform all the coupling reactions in low-protein binding tubes and all the assay reactions in low-protein binding, round-bottom, 96-well microtiter plates. Seal the plates with disposable adhesive foil or plastic 96-well microplate covers for the assay incubation steps. Use a magnetic plate separator to immobilize the beads during the assay wash steps.
NOTE: The dual-reporter flow analysis system has three lasers: (1) one that identifies and quantifies the bead set-specific fluorescence (Classification Channel); (2) one that detects and quantifies the target-specific phycoerythrin (PE) fluorescence (Reporter Channel 1; 532 nm excitation, "orange" 565-585 nm emission); and (3) one that detects and quantifies the target-specific BV421 fluorescence of a second target analyte (Reporter Channel 2; 405 nm excitation, "blue" 421-441 nm emission).
2. Coupling rhPD-1 to magnetic beads
NOTE: The protein to be coupled must be free of bovine serum albumin (BSA), sodium azide, glycine, glycerol, tris(hydroxy-methyl)aminomethane (Tris), or amine-containing additives and should be suspended in PBS at pH 7.4. A commercial coupling kit is available that includes all the necessary reagents and buffers described herein (see the Table of Materials).
- Remove all the coupling reagents from the refrigerator, and allow them to equilibrate to room temperature (RT, 18-22 °C) for 20-30 min.
- Resuspend the stock microspheres by briefly vortexing, sonicating, or rotating (15 min at 15-30 rpm), according to the product information sheet.
- Transfer 1 × 106 magnetic beads to a 1.5 mL low-protein binding microcentrifuge tube (see the Table of Materials).
- Wash beads with 100 µL of Activation Buffer15: 0.1 M NaH2PO4 (monobasic), pH 6.2.
NOTE: Coupling may also be performed using a pre-configured Coupling Kit, which includes 0.1 M 2-morpholinoethanesulfonic acid (MES), pH 6.0, as an alternative activation and coupling buffer (see the Table of Materials).- Place the tube containing the beads in a magnetic separator for 1-2 min.
NOTE: Alternatively, the beads can be separated by microcentrifugation at ≥8,000 × g for 1-2 min. - Aspirate the supernatant with a pipette from the magnet-immobilized or pelleted beads with the tube still positioned in the magnetic separator.
- Remove the microcentrifuge tube from the magnet, and add 80 µL of Coupling Buffer (see the Table of Materials).
- Vortex the reaction tube gently, and sonicate for 20 s to disperse the beads.
- Place the tube containing the beads in a magnetic separator for 1-2 min.
- Activate the beads with sulfo-NHS and EDC.
NOTE: The stock solution of sulfo-NHS is 50 mg/mL dissolved in Activation Buffer. The stock solution of EDC is also 50 mg/mL dissolved in Activation Buffer. Both the Activation Buffer and the moisture in the atmosphere cause EDC degradation. It is not advisable to use stored EDC solution. Make just enough fresh EDC solution before the step, and use it immediately when the solution is ready. Discard the excess EDC solution.
CAUTION: EDC causes severe eye irritation and is a respiratory tract and skin irritant.- Add 10 µL of sulfo-NHS to the microfuge tube containing the washed and activated beads.
- Add 10 µL of EDC stock solution to the microfuge tube containing the beads and sulfo-NHS.
- Protect the photosensitive microspheres from light, and rotate on the rotator for 20 min at 15-30 rpm, at RT (18-22 °C). Alternatively, the tube can remain stationary during the activation step if it is gently vortexed to redistribute the beads at 10 min intervals.
- Wash excess coupling reagents off the beads.
- Place the tube containing the activated beads in a magnetic separator for 1-2 min.
- Aspirate the supernatant with a pipette from magnet-immobilized or pelleted beads with the tube still positioned in the magnetic separator.
- Remove the microcentrifuge tube from the magnet, and add 100 µL of Activation Buffer.
- Vortex the reaction tube gently to disperse the beads.
- Repeat steps 2.6.1-2.6.4 two additional times for a total of three washes. At the end of washing, the beads will be suspended in 100 µL of Activation Buffer at an approximate concentration of 10 × 106 beads/mL.
- Couple the rhPD-1 peptide to the activated beads.
- Add 390 µL of Activation Buffer to the tube containing the activated beads to bring the total bead suspension volume up to 490 µL.
- Add 1 µg of PD-1 peptide to the activated bead suspension by adding 10 µL of PD-1 peptide solution (1 mg/mL dissolved in PBS) to the tube containing the activated beads.
- Briefly vortex the microcentrifuge tube to uniformly distribute the PD-1 and activated beads.
- Incubate the beads with PD-1 for 2 h in the dark at RT (18-22 °C) with rotation (15-30 rpm).
- Wash the beads twice (2x) using Assay/Wash Buffer (PBS-TBN: 1x PBS, pH 7.4 + 0.1% BSA + 0.05% Tween-20 + 0.05% NaN315).
- Place the tube containing the activated beads in a magnetic separator for 1-2 min.
- Aspirate the supernatant with a pipette from the magnet-immobilized or pelleted beads with the tube still positioned in the magnetic separator.
- Remove the microcentrifuge tube from the magnet, and add 100 µL of Activation Buffer.
- Vortex the reaction tube gently to disperse the beads.
- Repeat steps 2.8.1-2.8.4 one additional time for a total of two washes. At the end of washing, the beads will be suspended in 100 µL of Activation Buffer at a concentration of 10 × 106 beads/mL.
NOTE: The Assay/Wash buffer can be made without sodium azide (preservative) if the buffer is not also used as a storage medium. - Store the rhPD-1-coupled beads in the dark in the refrigerator at 2-8 °C if not used immediately. Protein-coupled beads are stable for up to 18 months.
3. Evaluation of successful rhPD-1 coupling to the beads
NOTE: The rhPD-1-coupled microspheres are reacted with biotinylated rhPD-L1, the latter of which is detected by incubation with SAPE followed by an assessment on the flow cytometer. This verifies both successful PD-1 coupling to the magnetic beads and also functional interaction between the rhPD-1 and rhPD-L1 proteins.
- Create a two-fold serial dilution series of biotinylated rhPD-L1 in PBS-TBN (the stock rhPD-L1 solution is 1 mg/mL). The final rhPD-L1 concentration range to be tested is an 8 µg/mL solution down to a 313 pg/mL solution. Create 150 µL volumes of each rhPD-L1 dilution: 50 µL for each reaction and two reactions per condition, plus sufficient excess to accommodate pipetting losses.
- Label the rhPD-L1 dilution microfuge tubes as 8 µg/mL, 4 µg/mL, 2 µg/mL, 1 µg/mL, 0.5 µg/mL, 0.25 µg/mL, 0.125 µg/mL, 0.063 µg/mL, and 0.031 µg/mL. A 0 µg/mL tube (PBS-TBN only) serves as the no-PD-L1 control.
- Pre-load 150 µL of PBS-TBN to all the labeled rhPD-L1 dilution tubes.
NOTE: The highest final rhPD-L1 concentration to be tested is 8 µg/mL, and the rhPD-L1 will be diluted 1:1 upon addition to the reaction mixture, so the dilution tube labeled "8 µg/mL" refers to the final concentration and actually contains 16 µg/mL rhPD-L1. The labels on all the dilution tubes indicate the final rhPD-L1 concentration after addition to the reaction. - Create the highest-concentration rhPD-L1 dilution (16 µg/mL actual). This is a two-step, 62.5-fold dilution of the 1 mg/mL rhPD-L1 stock solution (1,000 µg/16 µg = 62.5).
- Combine 84 µL of PBS-TBN with 16 µL of rhPD-L1 stock solution (1 mg/mL, i.e., 1,000 µL) in a microcentrifuge tube. This is a 6.25-fold dilution, and the resulting concentration is 160 µg/mL rhPD-L1.
- In the tube labeled "8 µg/mL", combine 270 µL of PBS-TBN with 30 µL of the rhPD-L1 dilution created in the previous step (160 µg/mL). This is a 10-fold dilution, and the resulting concentration is actually 16 µg/mL. The "8 µg/mL" tube label refers to its final concentration after addition to the reaction.
- Transfer 150 µL of the rhPD-L1 dilution created in step 3.1.3 ("8 µg/mL") to the "4 µg/mL" tube, close the microcentrifuge tube cap, and briefly vortex to mix the solution.
- Repeat step 3.1.4 sequentially until the rhPD-L1 dilution series is completed. After creation, all the tubes between "8 µg/mL" to "0.063 µg/mL", as well as the 0 µg/mL control, should contain 150 µL of solution, and the last tube, "0.031 µg/mL", should contain 300 µL of solution. This creates a sufficient volume of each dilution to test 50 µL of each biotinylated rhPD-L1 dilution in duplicate reactions, with sufficient excess remaining to accommodate pipetting losses.
- Count the rhPD-1-coupled beads using a hemocytometer17.
- Dilute the stock rhPD-1-coupled beads to 5 × 104 beads/mL, with a volume sufficient for 2,500 beads/50 µL/reaction.
- Vortex the 5 × 104 beads/mL rhPD-1-coupled bead suspension, and pipette 50 µL of the suspension into each labeled/mapped well of a 96-well round-bottom microtiter plate so that there are duplicate wells created for each rhPD-L1 dilution that is being tested.
- Add 50 µL of each biotinylated rhPD-L1 dilution tube created in step 3.1 to the appropriate wells on the microtiter plate.
- Cover the microtiter plate with a disposable foil or plastic adhesive plate sealer, and incubate the plate for 1 h in the dark at RT (18-22 °C) on an orbital shaker (600 rpm).
- Wash excess biotinylated rhPD-L1 from the beads.
- Transfer the sealed plate from the orbital shaker to the magnetic plate separator for 2 min to immobilize the beads.
- Carefully remove the adhesive plate sealer, confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatants into a sink or biohazard liquid waste container, as appropriate. Gently but briskly tap the inverted plate onto a cushion of absorbent paper tissue to remove the remaining supernatant.
- Remove the microtiter plate from the magnetic plate separator, and pipette 150 µL of PBS-TBN into each well.
- Place the unsealed plate on the magnetic separator for 2 min to immobilize the beads.
- Confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatants into a sink or biohazard liquid waste container, as appropriate. Gently but briskly tap the inverted plate onto a cushion of absorbent paper tissue to remove the remaining supernatant.
- Repeat the plate washing steps 3.7.3-3.7.5 two times for a total of three washes with 150 µL of PBS-TBN each. Ensure that no supernatant remains in the wells at the end of the last washing step. Work steadily to prevent the drying of the immobilized beads on the well bottoms.
- Add SAPE Detection Reagent.
- Dilute the SAPE stock solution in PBS-TBN to a working concentration of 6 µg/mL. Prepare a sufficient SAPE working solution volume so that all the reaction wells can receive 100 µL/well, with sufficient extra to accommodate pipetting losses.
- Remove the microtiter plate from the magnetic plate separator.
- Add 100 µL of SAPE working solution into each reaction well, and resuspend the washed beads by pipetting.
- Seal the 96-well microtiter plate with a foil or plastic adhesive plate sealer, and incubate for 1 h in the dark at RT (18-22 °C) on an orbital shaker at 600 rpm.
- Remove the microtiter plate from the incubator, transfer it to the magnetic plate separator to immobilize the beads, remove the adhesive plate sealer, and wash the beads three times with 150 µL PBS-TBN, as described in steps 3.7.3-3.7.5.
- After removing the final wash, remove the plate from the magnetic plate separator, and resuspend the beads in 100 µL of PBS-TBN per well.
- Analyze the results.
- Read the plate on the flow analysis instrument (see the Table of Materials) to determine the median fluorescence intensity (MFI) of each reaction using the following instrument settings: aspiration volume = 50 µL; minimum bead count = 100 beads; timeout setting = 40 s; gating = 7,000-17,000; operating mode = Single Reporter. Duplicate wells are run for each condition, and the two output MFI values for each condition are averaged before performing further data calculation and graphing.
NOTE: The MFI value of each dilution should show concentration-dependent binding, indicating acceptable rhPD-1 coupling efficiency to the beads and confirming good interaction of the recombinant PD-1/PD-L1 proteins.
- Read the plate on the flow analysis instrument (see the Table of Materials) to determine the median fluorescence intensity (MFI) of each reaction using the following instrument settings: aspiration volume = 50 µL; minimum bead count = 100 beads; timeout setting = 40 s; gating = 7,000-17,000; operating mode = Single Reporter. Duplicate wells are run for each condition, and the two output MFI values for each condition are averaged before performing further data calculation and graphing.
4. PD-L1 magnetic bead-based PD-1/PD-L1 blocking assay
NOTE: This assay assesses the blocking activity of soluble mediators (e.g., anti-PDL1-peptide antibodies) on recombinant PD1/PD-L1 interactions. Briefly, biotinylated rhPD-L1 is preincubated with antibodies generated in rabbits after different PDL1-Vaxx peptide inoculations. The rhPD-L1 + anti-PDL1 antibody mixture is then captured using rhPD-1-coupled magnetic beads, and the rhPD-L1 binding to the rhPD-1-coupled beads is quantified by the addition of streptavidin-PE. The PE fluorescence signal inversely correlates with the blocking activity of the tested anti-PDL1 antibodies/inhibitors. Anti-PDL1-peptide antibody binding is simultaneously assessed by the binding of a BV421-coupled anti-rabbit (for anti-PDL1-peptide antibodies) or anti-human (for control antibodies) secondary antibody and by evaluating the BV421 fluorescence in the instrument's second channel. The assay steps are pictorially detailed in Figure 1.
- Prepare a two-fold serial dilution series of the test antibodies, including PDL1-Vaxx-induced polyclonal antibody candidates, a negative control antibody (trastuzumab, Herceptin, humanized anti-HER2 monoclonal antibody), and a positive control antibody (atezolizumab; humanized anti-PDL1 IgG1 monoclonal antibody). Each reaction will use 25 µL of the assigned antibody dilution, so the volumes shown are sufficient for performing each reaction in duplicate wells per condition (i.e., 50 µL per condition), with some excess remaining to accommodate pipetting losses.
- For each vaccine-induced anti-PDL1-peptide antibody and control antibody, ensure that the range of the final antibody concentrations tested is from 1,000 µg/mL down to 8 µg/mL. Prepare stock solutions of all the antibodies at 2,000 µg/mL.
- For each antibody, label the dilution tubes as 1,000 µg/mL, 500 µg/mL, 250 µg/mL, 125 µg/mL, 63 µg/mL, 31 µg/mL, 16 µg/mL, and 8 µg/mL, and include the antibody name. For each antibody, also add a "0 µg/mL" tube, which will be the vehicle-only (PBS-TBN) control.
NOTE: When added to the reaction mixture, the antibody concentration will be diluted 1:1. The antibody dilution tubes are labeled as the final antibody concentration after addition to the reaction and actually contain twice the amount of antibody than labeled. - Add 75 µL of PBS-TBN to all the antibody dilution tubes labeled "500 µg/mL" and below, including the "0 µg/mL" vehicle-only control tubes.
- For each antibody, pipette 150 µL of the 2,000 µg/mL stock solution into the respective tube labeled "1,000 µg/mL". This will be used to make all the subsequent dilutions for each antibody.
- For each antibody, create a complete dilution series by pipette-transferring 75 µL from the "1,000 µg/mL" tube to the tube with the next lower dilution in the series (i.e., "500 µg/mL"). Close the newly completed dilution tube, vortex it briefly, and continue the construction of the dilution series by transferring 75 µL from the "500 µg/mL" tube to the "250 µg/mL" tube. Repeat this pattern until the last dilution, "8 µg/mL", has been made for all the antibodies.
NOTE: In the completed dilution series for each antibody, there should be a 75 µL volume for all the tubes except the lowest dilution, 8 µg/mL, which should contain a volume of 150 µL. Each reaction will use 25 µL of antibody dilution, so these volumes are sufficient for performing each reaction in duplicate wells per condition (i.e., 50 µL per condition), with extra to accommodate pipetting losses.
- Place 25 µL of the diluted antibodies into the designated wells of a 96-well microtiter plate.
- Dilute biotinylated rhPD-L1 to a working concentration of 4 µg/mL in PBS-TBN at a volume sufficient to include 25 µL in duplicate wells per condition (i.e., 50 µL per condition), with extra to accommodate pipetting losses.
NOTE: In this work, biotinylated rhPD-L1 at 4 µg/mL resulted in approximately 50% of the maximum MFI signal measured in the earlier coupling evaluation and was used for the PD-1/PD-L1 blockade analysis. - Add 25 µL of biotinylated rhPD-L1 (4 µg/mL) to each reaction well, cover the microtiter plate with a foil or plastic adhesive seal, and incubate at RT (18-22 °C) for 1 h with shaking on an orbital plate shaker at 600 rpm.
- Dilute the rhPD-1-coupled beads to 50,000 beads/mL, with a sufficient volume for 50 µL/well (2,500 beads/well) plus extra to accommodate pipetting losses.
- Remove the 96-well microtiter reaction plate from the shaker, and remove the adhesive plate seal.
- Add 50 µL of the rhPD-1-coupled bead mixture to each well.
- Seal the plate, and incubate for 1 h in the dark at RT (18-22 °C) on an orbital shaker at 600 rpm.
- Transfer the sealed plate from the orbital shaker to the magnetic plate separator for 2 min to immobilize the beads.
- Carefully remove the adhesive plate sealer, confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatants. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove excess supernatant.
- Wash excess reaction reagents from the beads.
- Remove the microtiter plate from the magnetic plate separator, and add 150 µL of PBS-TBN to each well.
- Place the microtiter plate on the magnetic plate separator for 2 min to immobilize the beads.
- Confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatants. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove excess supernatant.
- Repeat the plate washing steps 4.11.1-4.11.3 two times, for a total of three washes with PBS-TBN. Be sure the SAPE reagent is prepared (below) before removing the final (third) wash solution.
- Add the SAPE detection reagent.
- Dilute the stock SAPE stock solution to a 6 µg/mL working concentration in PBS-TBN; make a sufficient volume for 100 µL/well, plus extra to accommodate pipetting losses.
- Add 100 µL/well of SAPE working solution into each reaction well, and resuspend the beads by pipetting.
- Seal the plate, and incubate for 1 h in the dark at RT (18-22 °C) on an orbital shaker at 600 rpm.
- Decant the excess SAPE from the reaction.
- Transfer the sealed plate from the orbital shaker to the magnetic plate separator for 2 min to immobilize the beads.
- Carefully remove the adhesive plate sealer, confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatant. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove the supernatant containing excess SAPE.
- Wash the excess SAPE off the beads.
- Remove the microtiter plate from the magnetic plate holder.
- Add 150 µL of PBS-TBN to each well to resuspend the beads.
- Place the microtiter plate on the magnetic plate separator for 2 min to immobilize the beads.
- Confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatant. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove excess supernatant.
- Perform two additional wash steps with PBS-TBN for a total of three washes by repeating steps 4.15.1-4.15.4. Have the BV421-congugated secondary detection antibodies prepared (next step) before removing the final (third) wash solution.
- Add the BV421-congugated secondary antibodies.
- Dilute BV421-congugated anti-human IgG (for detecting humanized control antibodies) and BV421-congugated anti-rabbit IgG (for detecting PDL1-Vaxx-induced polyclonal antibodies) (see the Table of Materials) 1:400 in Wash/Assay Buffer at volumes sufficient for using 100 µL/well of each, with extra to accommodate pipetting losses.
- Add 100 µL of diluted BV421-conjugated anti-human IgG or BV421-conjugated anti-rabbit IgG to the appropriate wells.
- Seal the plate, and incubate for 1 h in the dark at RT (18-22 °C) on an orbital shaker at 600 rpm.
- Decant the excess BV421-conjugated secondary antibodies from the beads.
- Transfer the sealed plate from the orbital shaker to the magnetic plate separator for 2 min to immobilize the beads.
- Carefully remove the adhesive plate sealer, confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatant. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove the supernatant containing excess BV421-conjugated secondary antibodies.
- Wash the excess BV421-conjugated secondary antibodies from the beads.
- Add 150 µL of PBS-TBN to each well to resuspend the beads.
- Place the microtiter plate on the magnetic plate separator for 2 min to immobilize the beads.
- Confirm that the magnet and microtiter plate are securely together, and invert the plate and dump the supernatant. Gently tap the inverted plate onto a cushion of absorbent paper tissue to remove excess supernatant.
- Perform three additional wash steps with PBS-TBN for a total of four washes by repeating steps 4.19.1-4.19.3.
- After removing the last (fourth) wash buffer, remove the microtiter plate from the magnetic plate separator, and resuspend beads in 100 µL PBS-TBN/well with a pipettor.
- Analyze the results.
- Read the plate on the dual-reporter flow analysis system to determine the MFI of each reaction using the following instrument settings: aspiration volume = 50 µL; minimum bead count = 100 beads; timeout setting = 40 s; gating: 7,000-17,000; operating mode = Dual Reporter.
- With the dual-reporter system, ensure that Reporter Channel 1 measures the orange PE fluorescence (quantity of rhPD-L1 attached to rhPD-1-conjugated beads) and Reporter Channel 2 measures the blue BV421 fluorescence (quantity of attached blocking antibody bound to rhPD-L1).
- Run duplicate wells for each condition, and average the two output MFI values for each condition before performing further data calculation and graphing.
- Standardize the MFI value of each sample to the negative control, and calculate the percentage of inhibition for each sample:
Inhibition% = (100 × [Negative Control MFI − Sample MFI])/Negative Control MFI
NOTE: The negative control MFI values (no inhibition) are the highest values; the bound PD-L1 signal MFI is 100%, and the inhibition of rhPD-L1 binding to rhPD-1 is defined as 0%.
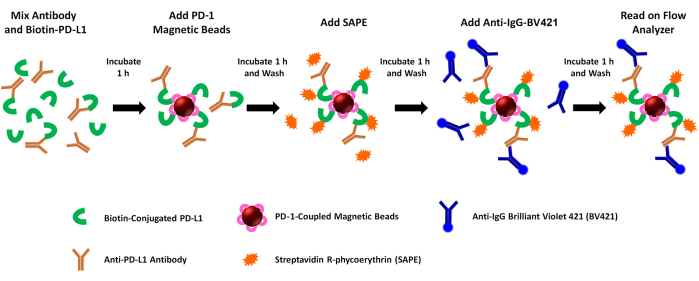
Figure 1: Schematic of the dual-reporter PD-1/PD-L1 blockade assay. Biotinylated recombinant human PD-L1 (rhPD-L1) is pre-incubated with selected PDL1-Vaxx-induced anti-PDL1 antibodies before combining with rhPD-1-coupled magnetic beads to allow PD-1/PD-L1 checkpoint complex formation. Complexed rhPD-L1 is then detected and marked by the addition of streptavidin-coupled phycoerythrin (SAPE, orange fluorophore). Antibodies against PDL1-Vaxx epitopes target rhPD-L1 that has complexed to rhPD-1 pre-coupled to the magnetic beads, and they are illuminated using a Brilliant Violet 421-conjugated secondary antibody (BV421, blue fluorophore). Both biotinylated rhPD-L1 that is complexed to PD-1 (PE signal) and anti-PDL1 antibodies that recognize and bind the rhPD-L1 (BV421 signal) are concurrently analyzed using a dual-reporter flow cytometric instrument that interrogates samples for both fluorophores in two separate reporter channels. The output values for each sample are the median fluorescence intensity of each fluorophore. The inhibition of PD1/PD-L1 complex formation by different PDL1-Vaxx-induced antibodies is then extrapolated by comparing the experimental signals to those generated using a negative control monoclonal antibody that does not bind rhPD-L1 (0% inhibition). Please click here to view a larger version of this figure.
The assay was able to precisely quantify the inhibition of the PD-1/PD-L1 interaction by four unique polyclonal antibodies, generated against the rhPD-L1 vaccine peptides, that are being explored as potential cancer therapeutic agents. The schematic of this assay is provided in Figure 1. The amount of biotinylated rhPD-L1 that bound to rhPD-1-conjugated beads and the inhibition of this binding by the four PLD1-Vaxx-induced antibody candidates was measured in Reporter Channel 1 using a streptavidin-PE detection reagent that directly bound rhPD-L1 (Figure 2).
All four polyclonal anti-PDL1-peptide antibodies blocked the rhPD-L1 interaction with PD-1 that had been immobilized on microspheres to variable extents. The percentage of inhibition of the different anti-PDL1 peptide antibodies ranged from 48% to 74% at the maximum tested concentration of 1,000 µg/mL. The positive control monoclonal antibody atezolizumab achieved 92% blockade of the PD-1/PD-L1 interaction at the maximum tested concentration14 of 4 µg/mL (Figure 2). All of the experimental PDL1-Vaxx antibodies showed concentration-dependent inhibition of rhPD-L1 binding to rhPD-1 conjugated beads compared with trastuzumab, the negative control antibody that was not expected to interact with the PD-1/PD-L1 system.
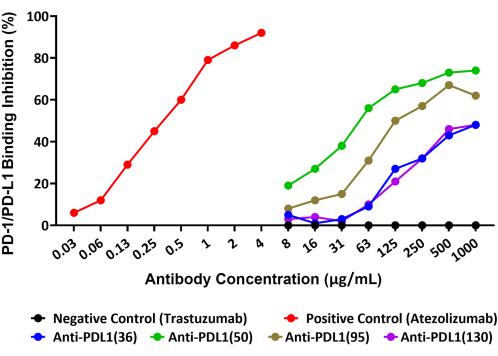
Figure 2: Blockade of rhPD-L1 interaction with rhPD-1 coupled to magnetic beads by anti-PDL1-peptide antibodies, as shown by a new fluorescent bead-based immunoassay. Recombinant human PD-1 was coupled to magnetic microspheres, and the beads were then incubated with biotinylated rhPD-L1 that had been pre-incubated with different anti-PDL1-peptide antibodies. A streptavidin-phycoerythrin detection reagent was used to bind the biotin and, thus, assess the relative quantity of rhPD-L1 that was available to bind to PD-1. Polyclonal antibodies raised in rabbits against the PDL1 peptide vaccines (anti-PDL1[36], anti-PDL1[50], anti-PDL1[95], and anti-PDL1[130]) were tested for inhibitory activity and showed 48%-74% blockade of recombinant PD-1/PD-L1 interactions at the highest concentration tested. Atezolizumab (a different anti-PDL1 monoclonal antibody) was used as the positive control. The unrelated commercial monoclonal antibody trastuzumab (anti-HER2) was used as a negative control. This figure is adapted from Guo et al.14. Please click here to view a larger version of this figure.
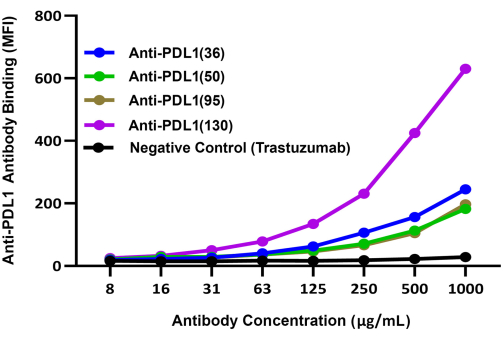
Figure 3: Comparative binding of different PDL1-Vaxx-induced antibodies to rhPD-L1 complexed to rhPD1-coated magnetic beads. Brilliant violet 421-conjugated secondary detection antibody was used to compare the binding of different rabbit polyclonal anti-PDL1-peptide antibodies to rhPD-L1 via rhPD-1 coated beads. The BV421 blue fluorescence signal was recorded in Reporter Channel 2 of the dual-reporter instrument; this signal correlates with the relative binding efficiency of the experimental anti-PDL1-peptide antibodies. Trastezumab (anti-HER2), a monoclonal antibody that targets a different checkpoint than PD-1/PD-L1, was used as a negative control. MFI represents the average bead median fluorescence intensity, which was measured in duplicate reaction wells per condition. This figure is adapted from Guo et al.14. Please click here to view a larger version of this figure.
The relative abilities of the four experimental PDL1-Vaxx-induced candidate antibodies to bind rhPD-L1 were compared using a separate detection system (BV421-conjugated anti-rabbit IgG) that was evaluated on the instrument's second reporter channel. These results indicated that all four polyclonal anti-PDL1-peptide antibodies bound to rhPD-L1 in a concentration-dependent manner14 (Figure 3). The anti-PDL1(130) antibody showed the highest rhPDL1 binding signal of the four PDL1-Vaxx-induced antibody candidates.
