1) Polytene chromosome slide preparation for laser capture microdissection
- Dissect half-gravid Anopheles females at 25 hr post-blood feeding. Fix ovaries from approximately five females into 500 µl of fresh modified Carnoy’s solution (100% methanol: glacial acetic acid, 3:1) at room temperature for 24 hr. Transfer ovaries to -20 °C for a long-term storage.
- Prepare Carnoy’s solution (100% ethanol: glacial acetic acid, 3:1) and 50% propionic acid just prior to making chromosome slides.
- Place one pair of ovaries in one drop of Carnoy’s solution on a Zeiss 1.0 PET membrane slide. Depending on the size, split ovaries into approximately 2-4 sections with dissecting needles and place them into a drop of 50% propionic acid on clean slides under a dissection microscope.
- Separate follicles and remove remaining tissue using paper towel under a dissection stereomicroscope. Add a new drop of 50% propionic acid to the follicles and allow them to sit for 3-5 min at room temperature.
- Place a siliconized coverslip on top of the droplet. Let the slide stand for approximately 1 min.
- Cover the slide with an absorbent material (filter paper is used for this method), and while using the eraser side of a pencil, apply a generous amount of pressure to the coverslip by tapping on it repeatedly with the eraser.
- Heat the slide to 60 °C on a slide denaturation/hybridization system for 15-20 min to aid in flattening the polytene chromosomes. Place slides into a humid chamber at 4 °C overnight to allow the acid to further flatten chromosomes.
- Place slides in cold 50% ethanol for 10 min. Gently remove the coverslip, and replace in cold 50% ethanol for 10 more min.
- Dehydrate slides in 70%, 90%, 100% ethanol for 5 min each. Air dry slides.
- Prepare a solution of GURR buffer solution by adding a single buffer tablet to 1 L of distilled water. Autoclave.
- Prepare the Giemsa solution by adding 1 ml of Giemsa staining solution to 50 ml of GURR buffer.
- Place air dried slides in Giemsa solution for 10 min and wash three times in 1X PBS. Air dry slides again in a controlled sterile climate to avoid contamination.
2) Laser capture microdissection of a single polytene chromosome arm
This section details the use of the PALMRobo software, which comes with the PALM MicroBeam Laser Microdissection system.
- Clean the microscope with 100% ethanol. Sterilize gloves and tubes with a UV light in a UV-crosslinker.
- Power up the PALM MicroBeam Laser Microdissection microscope and turn on laser. Open the laser dissection suite, PALMRobo, and configure the “Power” and “Focus” settings as necessary.
- Search for polytene chromosome arm of interest.
- Using the “Pencil” tool, outline the selected region.
- Open the “Elements window” from the menu bar.
- Select the “Drawn element,” ensure that you have selected “Cut.”
- Install the adhesive cap tube into the holder and place above the slide, leaving a small gap <1 mm in size, and start the laser cut.
- Place the “Catapult selection” within the cut site, leaving space between the edge and chromosome.
- Select “LPC” from the drop down option and begin catapulting.
- Check to ensure sample was catapulted into cap by pressing the “Eye” icon.
3) Purification of DNA from a single microdissected polytene chromosome arm
Follow the instructions of the QIAamp DNA Micro Kit to release and purify the collected DNA. Step 3.1 was modified to accommodate the inverted tube.
- Add 15 µl Buffer ATL and 10 µl proteinase K to the inverted tube (inside the cap) and incubate at 56 °C for 3 hr.
- Add 25 µl buffer ATL, 50 µl buffer AL, and 1 µl carrier RNA; mix. Add 50 µl 100% EtOH; mix.
- Transfer lysate to QIAamp column; centrifuge. Wash by adding 500 µl buffer AW1; centrifuge. Place column into new collection tube, add 500 µl buffer AW2; centrifuge. Place the column into a new tube; centrifuge to remove excess liquid.
- Place column into a 1.5 µl microcentrifuge tube and add 20 µl of water to elute; centrifuge.
- Evaporate freshly eluted DNA down to a final volume of 9 µl using a vacuumfuge.
4) Amplification of DNA from a single microdissected polytene chromosome arm
Two different protocols were used for the WGA of a single chromosome arm.
- DNA amplification and probe preparation via GenomePlex WGA
- Follow the GenomePlex Single Cell WGA4 Kit protocol to produce the first batch of amplified DNA:
- Add freshly prepared Proteinase K solution to the 9 µl sample; mix. Incubate DNA at 50 °C for 1 hr, then heat to 99 °C for 4 min. Keep on ice.
- Add 2 µl 1X Single Cell Library Preparation Buffer and 1 µl of Library Stabilization Solution; mix. Heat sample to 95 °C for 2 min. Cool on ice and centrifuge.
- Add 1 µl of Library Preparation Enzyme; mix and centrifuge. Incubate as follows:
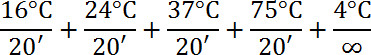
- Add 7.5 µl 10X Amplification Master Mix, 48.5 µl water. 5.0 µl WGA DNA Polymerase; mix and centrifuge.
- Thermocycle as follows:
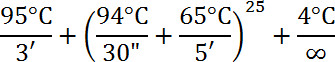
- Purify DNA using the Genomic DNA Clean & Concentrator Kit. The protocol is as follows:
- Add 5:1 DNA binding buffer:DNA sample (specifically for genomic DNA of less than 2 kb. If sample DNA is greater than 2 kb, use a 2:1 ratio) and transfer to provided spin column. Centrifuge.
- Add 200 µl DNA Wash Buffer and centrifuge. Repeat wash step. Then, add 50 µl of water and elute the DNA into a new 1.5 ml tube.
- Re-amplify sample DNA using the GenomePlex WGA3 Reamplification Kit as follows:
- Add 10 µl DNA to PCR tube (the kit recommends 10 ng total of DNA) with 49.5 µl water, 7.5 µl 10x Amplification Master Mix, 3.0 µl 10 mM DNTP mix, and 5.0 µl WGA DNA Polymerase. Mix and centrifuge.
- Use the following profile for the reaction:
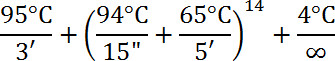
- Store DNA at -20 °C.
- Label DNA for FISH using the GenomePlex WGA3 Reamplification Kit as following:
- Create a master mix from the GenomePlex WGA3 Reamplification Kit by adding 10 µl DNA to PCR tube with 49.5 µl water, 7.5 µl 10X Amplification Master Mix, 3.0 µl 1 mM dNTP mix (1 mM of dATP, dCTP, dGTP, 0.3 µl 1 mM dTTP – if using labeled dUTP), 1 µl of 25 nM labeled dUTP, and 5.0 µl WGA DNA Polymerase.
- Use the following profile for the reaction:
- Ethanol precipitate the labeled probe by adding 1/10 the final reaction volume (7.5 µl for 75 µl reaction) of 3 M Sodium Acetate pH 5.2 and 2-3 volumes of 100% ethanol. Chill DNA sample at -80 °C for at least 30 min.
- Centrifuge sample at 4 °C for 10 min to create labeled pellet and remove supernatant and air-dry pellet.
- Create hybridization buffer as follows:
0.2 g Dextran Sulfate
1200 µl Deionized formamide
580 µl H2O
120 µl 20X SSC - Add 40 µl of hybridization buffer to air-dried pellet.
- Follow the GenomePlex Single Cell WGA4 Kit protocol to produce the first batch of amplified DNA:
- DNA amplification and probe preparation via REPLI-G Single Cell WGA followed by nick-translation
- Follow the REPLI-g Single Cell WGA Kit protocol to produce the amplified DNA:
- Prepare Buffer D2 (3 µl of 1 M DTT + 33 µl Buffer DLB).
- Mix 4 µl of purified microdissected material with 3 µl Buffer D2. Flick tube to mix. Incubate for 10 min at 65 °C. Add 3 µl of Stop solution; mix.
- Add 9 µl H2O, 29 µl REPLI-g Reaction Buffer, and 2 µl of REPLI-g DNA Polymerase to the sample. Incubate at 30 °C for 8 hr. Inactivate DNA polymerase by heating to 65 °C for 3 min. Store DNA at -20 °C.
- Follow the nick translation protocol to label REPLI-g amplified DNA:
- Prepare the following labeling mix:
1 µg of amplified DNA
5 µl 10X DNA Polymerase Buffer
5 µl 10X dNTP
5 µl 1X BSA
1 µl 1 mM Labeled dNTP
4 µl 1 U/µl DNase I
1 µl 10 U/µl DNA Polymerase I
H2O to 50 µl - Incubate at 15 °C for 2 hr. Add 2 µl of 0.5 M EDTA to stop reaction. Check DNA fragment size by running on gel.
- Follow steps from 4.1.4.3-4.1.4.6 to precipitate and solubilize pellet.
- Prepare the following labeling mix:
- Follow the REPLI-g Single Cell WGA Kit protocol to produce the amplified DNA:
5) 2D FISH on polytene and mitotic chromosome squash preparations
Please refer to detailed protocols for FISH on preparations of polytene squashes 47 and mitotic chromosome slides 48. Here we provide brief protocols.
- FISH on polytene chromosome squash preparations
- Immerse slides in 1X PBS for 20 min at RT. Fix slides in 4% Paraformaldehyde in 1X PBS for 1 min.
- Dehydrate slides through ethanol washes: 50%, 70%, 90%, 100% for 5 min each at RT. Air-dry slides.
- Prewarm the probe in hybridization buffer at 37 °C. Add 10-20 µl of probe to slide. Cover with 22 X 22 mm coverslip. Press out any air bubbles using a pipette tip.
- Denature chromosomes and probe at 90 °C for 10 min. Seal edges of coverslip with rubber cement. Transfer slide to humid chamber and incubate at 39 °C overnight.
- Wash the slides with 1X SSC at 39 °C for 20 min. Wash the slides with 1X SSC at RT for 20 min. Rinse slides in 1X PBS, then add Prolong anti-fade with DAPI. Cover with coverslip, and store in slide box at 4 °C for at least an hour before visualization.
- FISH on mitotic chromosome squash preparations
- Extract mitotic chromosomes from imaginal discs of 4th instar larva of An. gambiae.
- Prepare chromosome slides suitable for FISH.
- Add 2-3 µl of labeled DNA probe to hybridization buffer in a tube and mix gently by pipetting.
- Apply 10 µl of the probe mixture to the slide and cover with a 22 X 22 mm coverslip. Press out any air bubbles using a pipette tip.
- For counterstaining and detection, apply Prolong anti-fade with DAPI to the preparation and keep in dark for at least an hour before visualization.
6) 3D FISH on whole-mount ovarian tissue
- Prepare the following Buffer A mix:
60 mM KCl
15 mM NaCl
0.5 mM Spermidine
0.15 mM Spermine
2 mM EDTA
0.5 mM EGTA
15 mM PIPES - Prepare slides for nuclear visualization by adding a layer of nailpolish in a square pattern to match the size of the coverslips. This creates a raised surface to prevent squashing of nuclei when placing on coverslip in the future.
- Dissect fresh ovaries from Christopher’s stage 3 females and keep in a solution of 150-250 µl Buffer A with 0.5% digitonin. Run larger dissection needle over follicles (in tube with Buffer A with 0.5% digitonin) to destroy follicular membrane.
- Vortex for 5-10 min to further disturb follicles. Scrape down any large follicular pieces and centrifuge tube for 30 sec at lowest setting of ~500 revolutions per minute (RPM). Transfer supernatant to a new 2 ml Eppendorf tube, and add 100 µl of Buffer A. Repeat step 6.3 between 5-7 times, until the visible tissue is broken into small particles.
- Spin both tubes for 10 min at 2,000 RPM. Discard supernatant in both tubes. Note: Both tubes will be used for making final nuclear visualization slides. The tube with collected supernatant should contain primarily extracted nuclei, while the original tube with tissue will contain a mixture of tissue and nuclei embedded in nurse cells. Add 200 µl of Buffer A – 0.1% Triton and incubate overnight at 4 °C. Centrifuge 5 min at 10,000 RPM (10,621 x G) and remove supernatant.
- Add 200 µl 4% Paraformaldehyde in PBS. Incubate in thermomixer for 30 min, mixing at 450 RPM. Centrifuge 5 min at 5,000 RPM (2,655 x G) and remove supernatant. Wash with Buffer A with 0.1% Triton for 5 min, mixing at 450 RPM in thermomixer. Centrifuge 5 min at 5,000 RPM (2,655 G) and remove supernatant.
- Add pre-warmed at 37 °C labeled probe to tube. Denature at 95 °C in thermomixer, mixing at 450 RPM, for 10 min. Continue denaturation at 80 °C for 15 min with continued mixing. Incubate at 37 °C in thermomixer, with 450 RPM mixing overnight. Centrifuge 5 min at 5,000 RPM (2,655 x G). Remove supernatant.
- Wash with Buffer A with 0.1% Triton for 5 min. Centrifuge 5 min at 5,000 RPM (2,655 x G). Repeat 2 times. Apply drop of Prolong Anti-fade with DAPI.
- Pipet out nuclei/DAPI solution carefully (avoiding bubbles), apply to slide, and cover with coverslip.
Figure 1 describes the overall flow-through of the protocol described in this article. The user initially starts by microdissecting chromosome DNA samples from membrane slides. Microdissected material is extracted and purified. The purified DNA is then amplified, re-amplified, labeled, and then used for FISH to label chromosome spreads.
The LCM protocol can be broken down into three overall steps: 1) Finding chromosomes of interest and preparing the region for cutting (Figure 2A), 2) Cutting and catapulting the chromosome region of interest via laser (Figure 2B), and 3) Checking to determine if the sample is actually catapulted into adhesive cap (Figure 2C). The process of LCM used on the An. gambiae Sua strain polytene chromosomes is shown in Video 1. The video details the entire process from program start up, including important software functions and tips.
GenomePlex and REPLI-g single cell WGA kits used in this protocol differ greatly in resulting product size as well as overall yield. Figure 3 shows the results of gel electrophoresis ran for the GenomePlex and REPLI-g kits, as well as quantification of the samples by Nanodrop.
Microdissected material has been used to create FISH probes that target euchromatic regions of a chromosome. Figure 4 demonstrates FISH of five probes generated from microdissected material on polytene chromosomes of An. gambiae. Four autosomal arms were labeled with three fluorophores using the WGA3 kit: the 3R chromosome is labeled in green (fluorescein), the 3L chromosome is in a mixture of red (Cy-3) and yellow (Cy-5), the 2R chromosome is in yellow (Cy-5), and the 2L chromosome is in red (Cy-3). The X chromosome was labeled in orange (Cy-3) using nick-translation of the REPLI-g material in a separate experiment. To establish the correspondence between euchromatic portions of polytene and mitotic chromosome arms, chromosome paints have been hybridized to interphase, prophase, prometaphase and metaphase chromosomes of An. gambiae (Figure 5). To visualize the 3D organization of a single polytene chromosome arm in the cell nucleus, whole mount FISH was performed on An. gambiae Sua strain ovarian nurse cells (Video 2). Distinct chromosome arm territories are clearly seen in nuclei with interphase (Figure 5D) and polytene (Video 2) chromosomes.
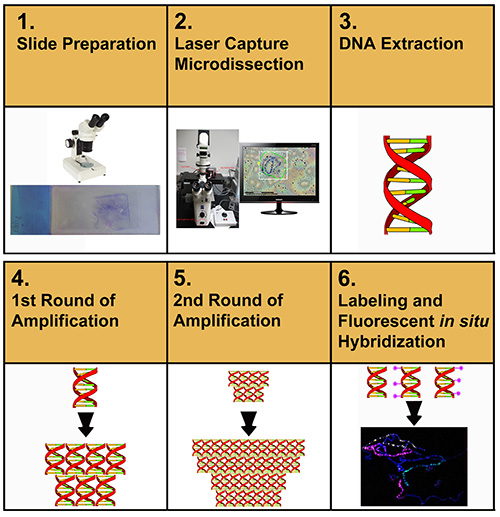
Figure 1. Schematic representation of the experimental procedures toward the preparation of chromosome paints. Click here to view larger image.
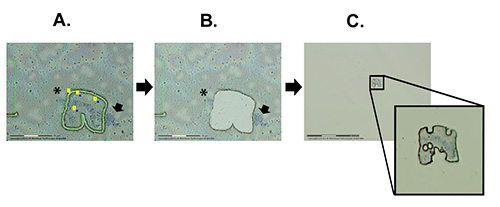
Figure 2. The major steps in chromosome microdissection. A) Laser-assisted cutting of the chromosomal region of interest through the membrane. B) The membrane with a hole after the catapulting is performed. C) The view of the catapulted piece of the membrane with a chromosomal segment in it attached to the adhesive cap. The arrow indicates the heterochromatin of the X chromosome that remained on the slide. The asterisk shows a piece of another chromosome that remained on the slide. Click here to view larger image.

Figure 3. Agarose gel images showing DNA after WGA. A) Low molecular weight (200-500 bp) DNA from arm 3R after using the WGA4 and WGA3 GenomePlex kits. B) High molecular weight (10-20 kb) DNA from arm 2L after REPLI-g amplification. The 100 bp ladder is shown in the left lanes. The tables below gel images show DNA concentrations measured by Nanodrop. Click here to view larger image.
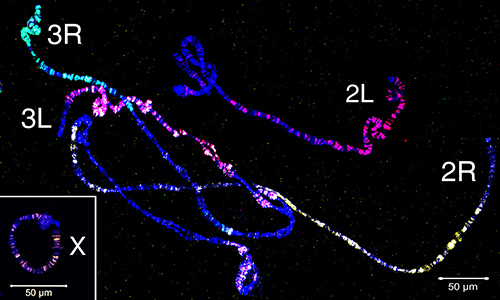
Figure 4. Painting of polytene chromosomes from ovarian nurse cells of An. gambiae using four probes generated from microdissected material. The X chromosome is labeled in orange (Cy-3) by nick-translation of the REPLI-g material. The 2R arm is labeled in yellow (Cy-5); the 2L arm is in red (Cy-3); the 3R arm is labeled in green (fluorescein); the 3L arm is labeled in a mixture of red (Cy-3) and yellow (Cy-5). Autosomes are labeled with the WGA3 amplification kit. Chromatin is stained in blue (DAPI). Chromosome names are placed near telomeric regions. Click here to view larger image.
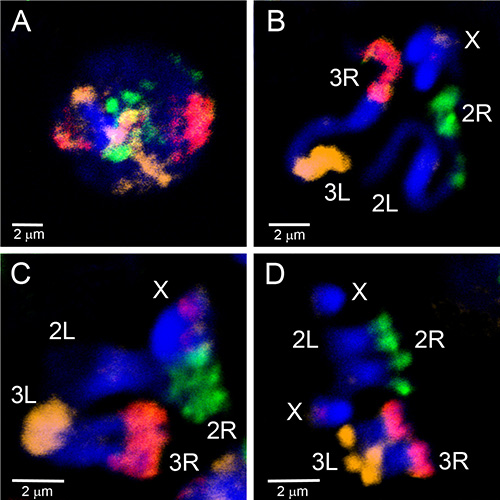
Figure 5. Painting of interphase (A), prophase (B), prometaphase (C) and metaphase (D) chromosomes from larval imaginal discs of the An. gambiae Mopti strain using three probes generated from microdissected material labeled by WGA3. The 2R arm is labeled in green (fluorescein); the 2L arm is unlabeled; the 3R arm is in pink, a mixture of red (Cy-3) and orange (Cy-5); the 3L arm is labeled in orange (Cy-5). The X chromosome has a red label corresponding to the 18S rDNA probe. Chromatin is stained in blue (DAPI). Brightly stained regions of chromosomes correspond to the heterochromatin. Click here to view larger image.
Video 1. The process of LCM of the An. gambiae polytene chromosomes. Click here to view Video 1.
Video 2. Whole mount 3D FISH performed on An. gambiae ovarian nurse cells. The probe is labeled in Cy-3 (depicted in blue) and was made from a microdissected 2R chromosome arm. Chromatin was stained with DAPI and is depicted by cyan pseudo-coloring (light blue). Click here to view Video 2.
