This experimental protocol describes how to image cell division in C. elegans embryos during mid embryogenesis. In particular, it describes how to image neuroblasts, which may facilitate epidermal morphogenesis. Epidermal morphogenesis occurs due to a combination of epidermal cell shape changes, migration and adhesion, but also relies on chemical or mechanical cues from the underlying neuroblasts (Figure 1B). The neuroblasts secrete guidance cues that are received by receptors on the surface of ventral epidermal cells, which regulates their migration10,14,25. Furthermore, the neuroblasts are highly proliferative, which may provide mechanical forces that contribute to the migration of ventral epidermal cells26. This protocol can be used to study neuroblast cell division during mid embryogenesis. First, embryos coexpressing markers to visualize neuroblasts (HAM-1:GFP) undergoing cytokinesis (mCherry:PH) are generated. HAM-1:GFP expresses GFP (green fluorescent protein) tagged protein that localizes to neuroblast nuclei27, and is necessary to use since there are many other cell types present during mid embryogenesis (>300 cells; Figure 3). This also acts as a good marker for dividing cells, since DNA condenses into chromosomes during mitosis. mCherry:PH is an mCherry-tagged fluorescent protein that is expressed by a maternal promoter (pie-1), and contains the lipid binding domain from PLC1∂1 28, which localizes to cell membranes. This protein is needed to visualize cytokinesis, when the membrane and cytosol pinch in to separate the mother cell into two daughter cells; Figure 3; Video 1). Although this protocol describes how to image neuroblast division, (shown by HAM-1:GFP expression), other markers could be used to visualize other cell types.
To visualize dividing neuroblasts, embryos coexpressing HAM-1:GFP and mCherry:PH are imaged every 2 min (for a total of 10 min). Each neuroblast is only ~1-4 μm in diameter, and is <1 μm thick, and it is best to use an objective with high magnification (e.g. 100X). Furthermore, numerous Z-stacks (~20) are collected with a small step size (0.2 μm) to ensure that the various angles of each cell (spherical in shape) are imaged. To collect this many images without compromising time points, a highly motorized stage (e.g. Piezo Z Stage) is recommended. After collecting a series of images, they are analyzed to find cells within a few Z-stacks that are dividing , the DNA is condensed, the membrane is ingressed and the new daughter cells are forming (time points of the projected stack of merged channels are shown in Figure 3A, and selected stacks of merged channels are shown in Figure 3B; Video 1). To better visualize the cells, it is recommended to keep each channel in grayscale and invert the images (e.g. Figure 4).
The image resolution is different for the widefield system using a high resolution CCD camera (1344 x 1024 pixels) vs. the swept field system using a high speed EMCCD camera with high sensitivity (~35 frames/sec and 512 x 512 pixels). Inverted images of dividing neuroblasts from control embryos taken with both systems are presented for comparison (Figure 4 vs. 5). As shown in Figure 4A (widefield system; Video 2), the images are clearer vs. Figure 5A (swept field system; Video 4). However, when using the widefield system, embryos are susceptible to phototoxicity and faster time points are not possible with the high resolution camera. For example, to examine changes in the localization of various proteins (e.g. to study changes in their dynamics), time points might need to be collected more often. Also, it may not be possible to image for longer periods of time without reducing the number of Z-stacks and time points. Using an EMCCD camera on the widefield system may permit a wider range of imaging applications. Cameras that have both high resolution and high speed have been developed, but the drivers may not be available depending on the acquisition software being used, and they still may not be as sensitive as EMCCD cameras. Another system that is commonly used for imaging C. elegans is the spinning disk confocal, which also has lower phototoxicity and can be outfitted with either EMCCD or CCD cameras (see Discussion).
To study genes that are required for cell division, hermaphrodites are treated with RNAi against a gene of interest (e.g. ani-1; see Sections 1.2 and 3 of the protocol) and their embryos are imaged in the same way as control embryos. To compare cells from RNAi embryos to control embryos, it is best to image at similar stages of embryonic development (to help match cell shapes and positions). For example, there is a larger cell visible in the center of the embryo during ventral enclosure that can act as a good reference point for imaging neuroblasts (Figures 4 and 5). The gene studied here, ani-1 (anillin), organizes actomyosin contractility, but is not required for cytokinesis in the early embryo23,29-30. Anillin’s homologues, however, are required for cytokinesis in higher eukaryotes16, and ani-1 is required for neuroblast cytokinesis (Fotopoulos, Wernike and Piekny, unpublished observations). As shown in Figures 4B (Video 3) and 5B (Video 5), several neuroblasts initiate cytokinesis and their membranes pinch in, but instead of forming two separate daughter cells, their membranes regress and the cells become multinucleate (>1 nucleus/cell). It must be noted that ani-1 may be required for the division or shape change of other cell types, which are not revealed by this protocol. Furthermore, this protocol does not show results from tissue specific RNAi (see Discussion).
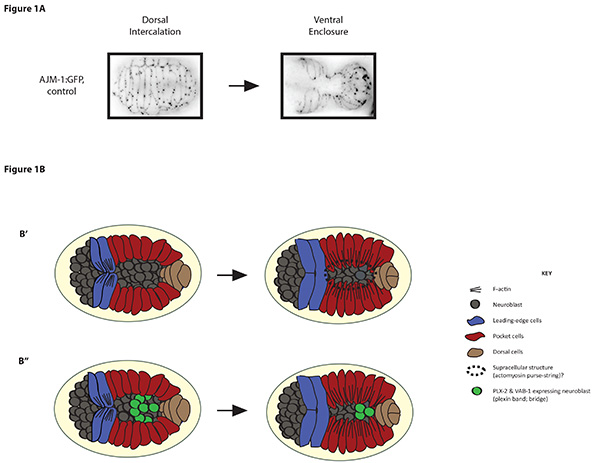
Figure 1. Ventral enclosure during C. elegans epidermal morphogenesis. A) Z-stack projections (3 Z-stacks of 0.5 μm in thickness) of a control AJM-1:GFP (adherens junction marker to visualize epidermal cell boundaries; strain ID at CGC SU159) embryo undergoing dorsal intercalation (left, dorsal view) and a control AJM-1:GFP embryo undergoing ventral enclosure (right, ventral view). Images are inverted to better visualize cell boundaries. B) Cartoons show ventral views of C. elegans embryos undergoing ventral enclosure. First, two pairs of leading edge cells (blue) use actin rich filopodia (black lines) to migrate toward the ventral midline (embryo on the left). Next, the posterior ventral pocket cells (red) migrate toward the midline creating a hole on the ventral surface that may close by a purse string like mechanism (B’; black dotted line/circle; reminiscent of wound healing)25. Also shown are the underlying neuroblasts (as grey circles). The second embryo (B”) shows how the migration of specific subsets of epidermal cells are mediated by a ‘bridge’ formed from the rearrangement of PLX-2/plexin and VAB-1/Ephrin receptor-expressing ‘plexin band’ cells (green circles). Click here to view larger image.
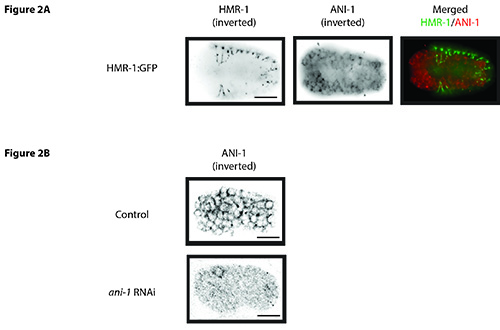
Figure 2. ANI-1 localization in C. elegans embryos. A) A fixed C. elegans embryo expressing HMR-1:GFP (E-cadherin; marker of epidermal cell boundaries) costained for GFP (green) and ANI-1 (red). Confocal images of the outer cell layers (4 Z-stacks of 0.2 μm in thickness) show the overlying epidermal cells and underlying neuroblasts, which are ANI-1 positive. B) Fixed N2 and N2; ani-1 RNAi embryos are co stained for ANI-1. ANI-1 is greatly reduced in the ani-1-depleted embryo. Scale bars: 10 μm. Click here to view larger image.
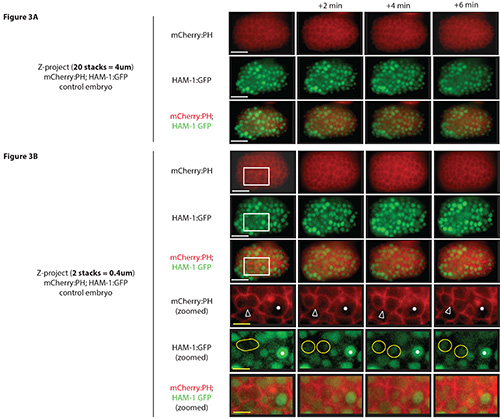
Figure 3. Visualizing dividing neuroblast during C. elegans embryogenesis. (A) Z-stack projections (20 Z-stacks of 0.2 μm in thickness) are shown from a control embryo coexpressing HAM-1:GFP (to visualize neuroblast nuclei; green) and mCherry:PH (to visualize cell membranes; red). Many neuroblasts and other cell types are visible in the projections. (B) Z-stack projections using a smaller number of Z planes are shown from another control embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). An area is zoomed in (white box) to more clearly show an individual dividing neuroblast. White arrow heads point to the ingressing plasma membrane of a dividing cell and green circles highlight the condensed DNA during mitosis and newly forming daughter nuclei towards the end of mitosis. The large cell serves as a reference point (white asterisk) to determine developmental stage and location within the embryo. Scale bars: 10 μm (white) and 3.5 μm (yellow). Click here to view larger image.
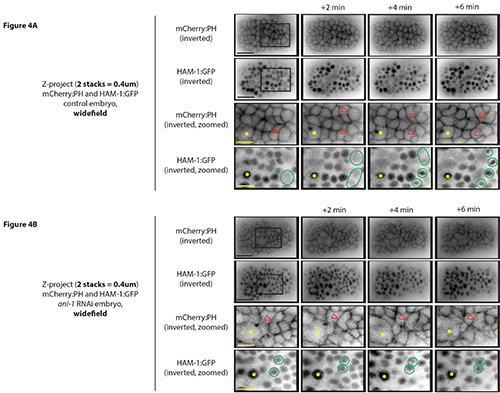
Figure 4. Visualizing neuroblast cytokinesis during C. elegans embryogenesis using a widefield system. A) Inverted Z-stack projections (2 Z-stacks of 0.2 μm in thickness) are shown from a control embryo coexpressing HAM-1:GFP and mCherry:PH collected from the widefield system using a CCD camera with high-resolution. Red arrowheads point to dividing neuroblasts with ingressing cell membranes. Green circles highlight condensed DNA during early phases of mitosis or newly forming daughter nuclei towards the end of mitosis/cytokinesis. The large cell serves as a reference point (yellow asterisk) to determine developmental stage. B) Images shown are similar to A), but are from an embryo treated with ani-1 RNAi. The cell membrane ingresses in as the cell proceeds through mitosis, but relaxes shortly after, leaving a multinucleate cell. Scale bar: 10 μm (black), 3.5 μm (yellow). Click here to view larger image.
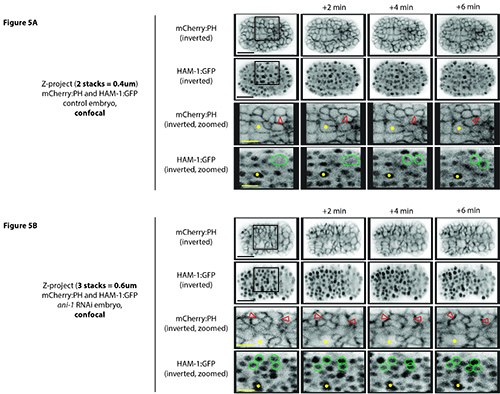
Figure 5. Visualizing neuroblast cytokinesis during C. elegans embryogenesis using a swept field system. A) Inverted Z-stack projections of 2 Z-stacks of 0.2 μm (total 0.4 μm) are shown from an embryo coexpressing HAM-1:GFP and mCherry:PH collected from the swept field system using an EMCCD camera with high sensitivity, but lower resolution. The red arrowhead points to a neuroblast undergoing cytokinesis. Green circles highlight condensed DNA during mitosis and the newly forming daughter nuclei after cytokinesis. The large cell serves as a reference point (yellow asterisk) to determine developmental stage. B) Images shown are similar to A), but are from an embryo treated with ani-1 RNAi, and 3 Z-stacks are used (total 0.6 μm). As described above, the membrane ingresses in as the cell proceeds through mitosis, but then relaxes causing the cell to become multinucleate. Scale bar: 10 μm (black), 3.5 μm (yellow). Click here to view larger image.
Video 1. Visualizing dividing neuroblasts during C. elegans embryogenesis. Z-stack projections of two Z planes (0.4 μm/plane) from a control embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). Images collected from the widefield microscope are shown for both channels. The video corresponds to the embryo shown in the third panel of Figure 3B. Click here to watch video.
Video 2. Visualizing dividing neuroblasts during C. elegans embryogenesis. Z-stack projections of two Z planes (0.4 μm/plane) from a control embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). The video corresponds to inverted images collected from the widefield microscope for the mCherry:PH channel only (embryo in Figure 4A). Click here to watch video.
Video 3. Visualizing dividing neuroblasts during C. elegans embryogenesis. Z-stack projections of two Z planes (0.4 μm/plane) from an ani-1 RNAi treated embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). The video corresponds to inverted images collected from the widefield microscope for the mCherry:PH channel only (embryo in Figure 4B). Click here to watch video.
Video 4. Visualizing dividing neuroblast during C. elegans embryogenesis. Z-stack projections of two Z planes (0.4 μm/plane) from a control embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). The video corresponds to inverted images collected from the swept field confocal microscope for the mCherry:PH channel only (embryo in Figure 5A). Click here to watch video.
Video 5. Visualizing dividing neuroblast during C. elegans embryogenesis. Z-stack projections of three Z planes (0.4 μm/plane) from an ani-1 RNAi treated embryo coexpressing HAM-1:GFP (green) and mCherry:PH (red). The video corresponds to inverted images collected from the swept field confocal microscope for the mCherry:PH channel only (embryo in Figure 5B). Click here to watch video.
