Figure 1 shows an absorption spectrum of a dry film of bR deposited on the surface of the diamond internal reflection element used for ATR spectroscopy. Characteristic bands from vibrations of the peptide bond (amide A, amide I and amide II) are clearly distinguishable. The approximate thickness of the dry film can be estimated as ~1 μm, considering the amount of added protein (18 μg) and the surface of the ATR (~0.2 cm2), and taking the density of protein as ~1.4 g/cm3,58 and of lipids as 1.0 g/cm3,59 and a lipid/protein ratio of 1/3 (w/w) in purple membrane60.
The dry film was rehydrated with excess of 4 M NaCl, 100 mM NaPi at pH 7.4 (Figure 3). The hydration level and effective protein concentration in the volume probed by the evanescent wave can be deduced from the scaling factor needed to digitally remove absorption bands from water. The optimum scaling factor was 0.87, meaning that in this case the buffer occupies 87% and the sample 13% of the volume near the surface. Taking into account protein and lipid density and the lipid/protein ratio in purple membranes (see above), we can deduce an effective protein concentration of 125 mg/ml in the volume probed by the evanescent wave. We can deduce from the hydration level that, in this particular case, the sample film thickness expanded ~6 times upon rehydration, from ~1 to ~6 μm. For a 45° incident angle, typical for our and for most ATR arrangements, the penetration depth of the evanescent field (dp) for a hydrated protein film varies between 0.3 and 0.6 μm in the 1,800-850 cm-1 interval 61. Because the evanescent field is almost insensitive to the sample located two times above dp 61, we infer that the amount of protein used in the ATR experiment could be in principle reduced 5x without any significant loss of signal.
When using buffers of lower ionic strength the film expands more, and the amount of protein in the volume probed by the evanescent field is reduced (Figure 2). The exact dependence between film swelling and the ionic strength of the buffer will depend, among other factors, on the nature of the lipids. For instance, a similar film swelling as obtained here for bR in purple membrane using 4 M NaCl was obtained for a membrane protein reconstituted in polar E. coli lipids using just 0.1 M NaCl 62. If the sample occupies less than 5% of the probed volume consider re-doing the hydration step using a buffer of higher ionic strength. Film swelling after hydration requires some time to reach stabilization (Figure 4). For bR in purple membrane the process is monoexponential, with a time constant of 12 min: it takes no more than 30-60 min to have a stable rehydrated film
Figure 5 shows an IR absorption spectrum of a hydrated film of ChR2 obtained by transmission. Here, hydration was achieved by exposing the dry film to an atmosphere of controlled humidity provided by a mixture of glycerol/water. The spectrum can be decomposed into contributions from water, detergent and protein. We could estimate the amount of each of them using extinction coefficient spectra, scaled to fit the experimental spectrum. For water we took the extinction coefficient spectrum from the literature63, and for DM we measured it from a 100 mg/ml solution (Figure 6). We also used extinction coefficients for two representative membrane proteins: the mitochondrial ADP/ATP carrier64 and bR (Figure 7). The scaling factor indicates a water and DM mass surface density of 260 μg/cm2 and 200 μg/cm2 in the film, respectively. The remaining absorption (Figure 5, red line) comes mainly from the protein, estimated to be at a surface density of 250 μg/cm2 using the amide II extinction coefficient from two different membrane proteins (see Figure 6). The molar ratio for protein/detergent/water was calculated to be 1/60/2000 using a molecular mass of 35 kDa, 480 Da, and 18 Da, respectively.
A 3D plot from typical time-resolved step-scan FT-IR experiment on bR is presented in Figure 10A, obtained by ATR and involving 200 min of data acquisition. Spectra can be extracted at specific times, for instance when the L, M and N intermediates of the bR photocycle are expected to reach their highest population (Figure 11). Their spectral features have been described extensively13,41,65 and will be not discussed further here. Figure 12 shows time-traces at some selected wavenumbers. Namely, the rise of the kinetics at 1,762 cm-1 (t1/2 ~60 μsec) reports on the dynamics of Asp85 protonation from the retinal Schiff base66, and its decay to zero indicates its deprotonation upon ground-sate recovery67. The negative rise of the kinetics at 1,740 cm-1 (t1/2 ~1 msec) reports on the deprotonation of Asp96 side chain, the proton donor to the Schiff base68. The decay of the intensity to zero reports on its reprotonation from the cytoplasm67. The dynamics of protein conformational changes can be probed by absorption changes in the amide I and amide II region, which reaches a maximal change at ~3 msec at room temperature67,69.
The application of SVD to spectroscopic problems has been reviewed before55,56. Briefly, SVD factorizes the experimental data, arranged into a matrix A, as: A = U S VT. This factorization can be modified to account for the noise dependence on wavenumber and to penalize fluctuations/drifts in the baseline57. The columns of U and V contain orthonormal spectra and time-traces vectors for each SVD component, respectively. S is a diagonal matrix containing the so-called singular values. The (abstract) spectro-temporal components in U and V appear in decreasing relevance to describe the experimental data in the least-square sense, quantified by their associated singular value. Figure 13A shows the singular values as a function of the component number, and reproduces the first eight columns/components of U (abstract spectra, Figure 13B) and V (abstract time-traces, Figure 13C). The signal is concentrated in the first five components (as expected for a photocycle containing five intermediate states), with components above largely dominated by random noise and other sources of errors. The experimental data was reconstructed using only the first five columns of U and V, and the first five columns and rows of S. The data reconstructed by SVD represents the best least-square approximation of the experimental data to a matrix of rank five. The reconstructed data shows improved quality (Figure 10B). Namely, the noise is largely reduced, as well as some barely noticeable fluctuations and drifts in the baseline (common in time-resolved step-scan spectroscopy 25). SVD processing is especially beneficial for improving the quality of the experimental time-traces in the millisecond range (red lines in Figure 12).
Time-resolved step-scan data for the ChR2 photocycle were obtained using the above described protocol for transmission experiments (Figure 14A), roughly involving 120 hr of accumulated measurements. The time-resolved data collected by step-scan extends from 6.25 µsec to 125 msec. The photocycle of ChR2 requires approximately 60 s for full recovery. The latest part of the photocycle can be covered using rapid-scan FT-IR, and both step-scan and rapid-scan data sets merged as presented elsewhere48. The spectral changes of ChR2 are ~10-fold smaller than those obtain from bR, making the measurements more challenging. Specially, oscillations in the baseline in the millisecond range become clearly evident at these low absorption changes. These are due to small oscillations in the mobile mirror in the millisecond time scale25. The oscillations, together with part of the noise, can be largely removed by SVD, remarkably improving the appearance of the data (Figure 14B).
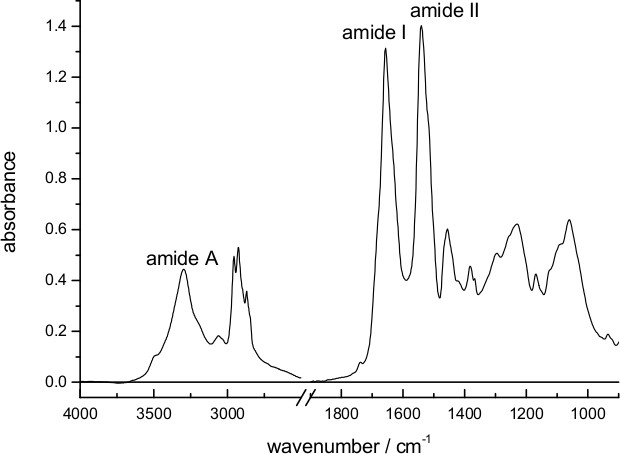
Figure 1. Absorption spectrum of bR in purple membrane dried on top the diamond surface of an ATR accessory. Bands from vibrations of the peptide bond (amide I, II, and A) are indicated.
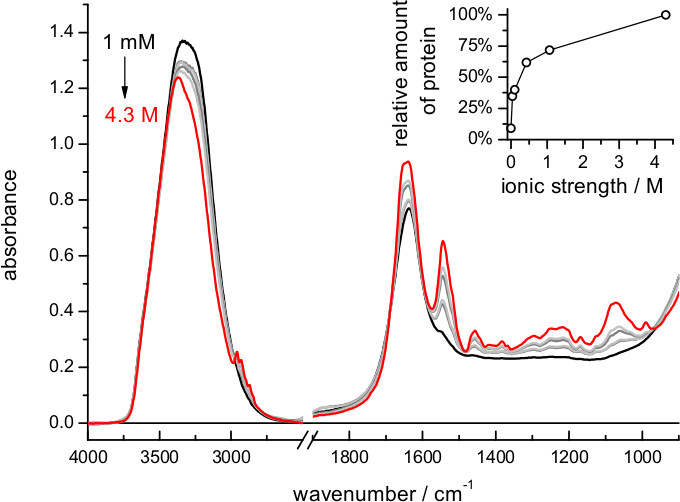
Figure 2. Absorption spectrum of a film of bR after rehydration using buffers of various ionic strengths (dilutions of 4 M NaCl, 100 mM Na2HPO4/NaH2PO4 at pH 7.4). Notice the decrease of the amide II band, i.e., increased film swelling, with decreasing ionic strength of the buffer. (Insert) Relative amount of protein near the surface, quantified by the intensity of the absorbance at 1,541 cm-1 (amide II maximum) after subtraction of the absorption contribution of the buffer.
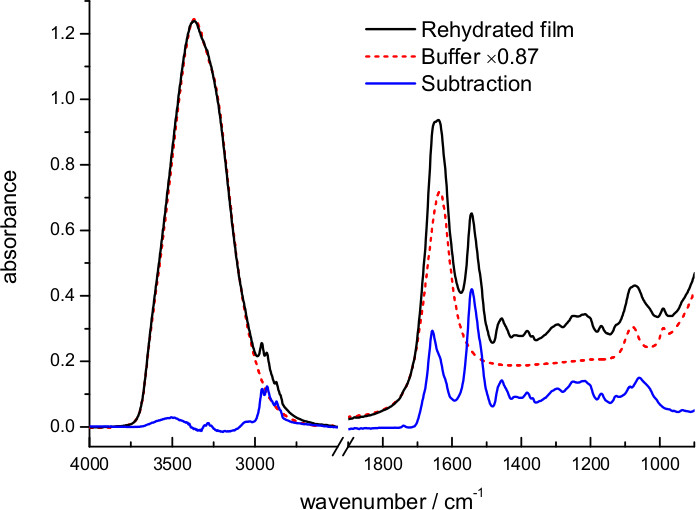
Figure 3. Absorption spectrum of a film of bR rehydrated with bulk buffer (4.3 M ionic strength) and after subtraction of the buffer contribution. The subtraction factor, 0.87, was chosen to remove the strong water absorption between 3,700-3,000 cm-1 and to obtain a flat baseline between 2,700-1,800 cm-1.
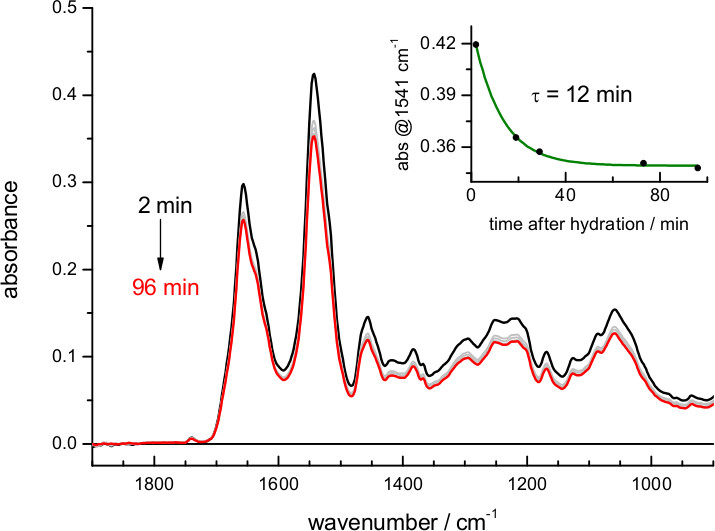
Figure 4. Absorption spectrum of bR after rehydration with a buffer of 4.3 M ionic strength (buffer absorption has been subtracted for clarity as in Figure 3). The insert shows the evolution of the film swelling after rehydration, followed by the protein amide II absorbance at 1,541 cm-1. A fit to a single exponential indicates a time constant for film swelling stabilization of 12 min.
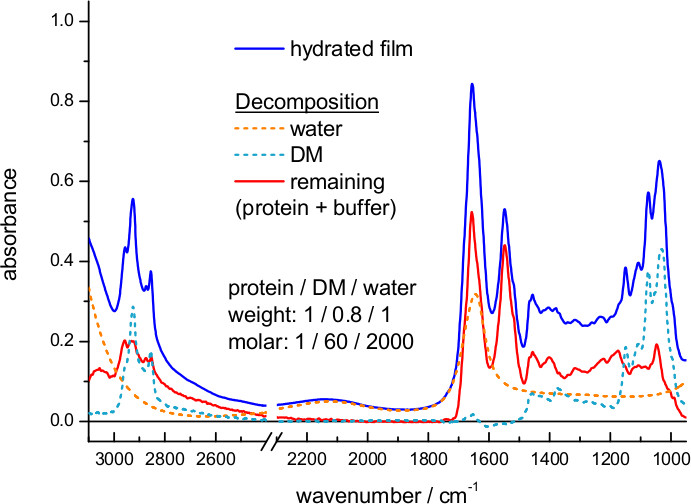
Figure 5. Absorption spectrum of a hydrated film of ChR2 measured by transmission (blue line). The extinction coefficient spectrum of water (dashed orange line) and DM (dashed cyan line) was scaled and subtracted (red line).
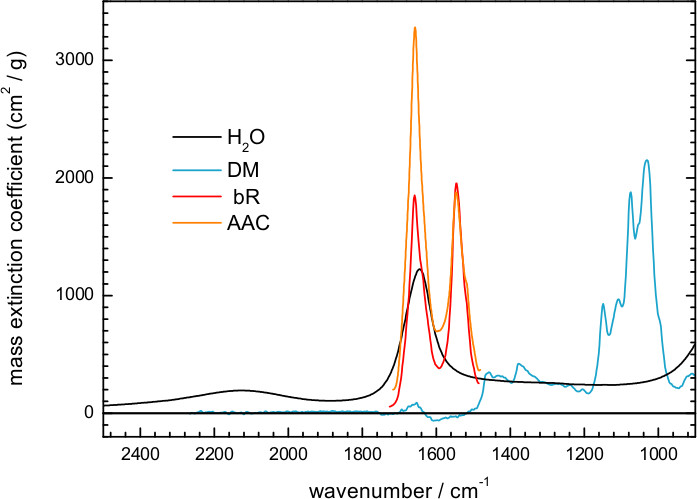
Figure 6. Reproduced mass extinction coefficient spectra at 25 °C for liquid water (http://www.ualberta.ca/~jbertie/JBDownload.HTM#Spectra) and for a hydrated film of the ADP/ATP carrier (AAC)64. The mass extinction coefficient spectrum was measured in solution for DM (100 mg/ml), and in a hydrated film for bR.
Figure 7. Transmittance of the optical filter used in the time-resolved scan-scan FT-IR measurements.
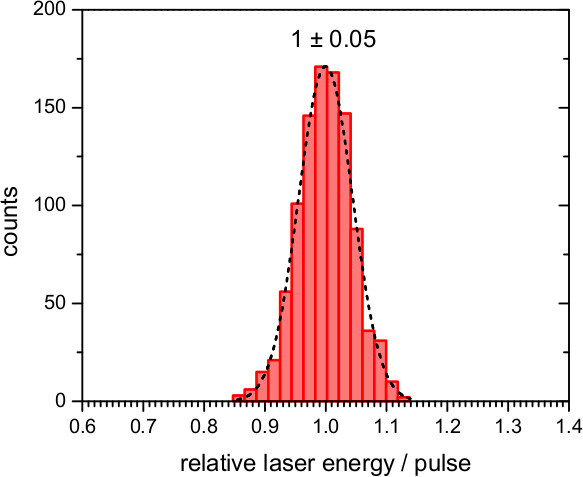
Figure 8. Performance of the laser pulses provided by the second harmonic (532 nm) of a Nd:YAG laser. Histogram of the variation of the relative energy of 1,000 laser pulses, and fit to a Gaussian distribution with a standard deviation of 0.05.
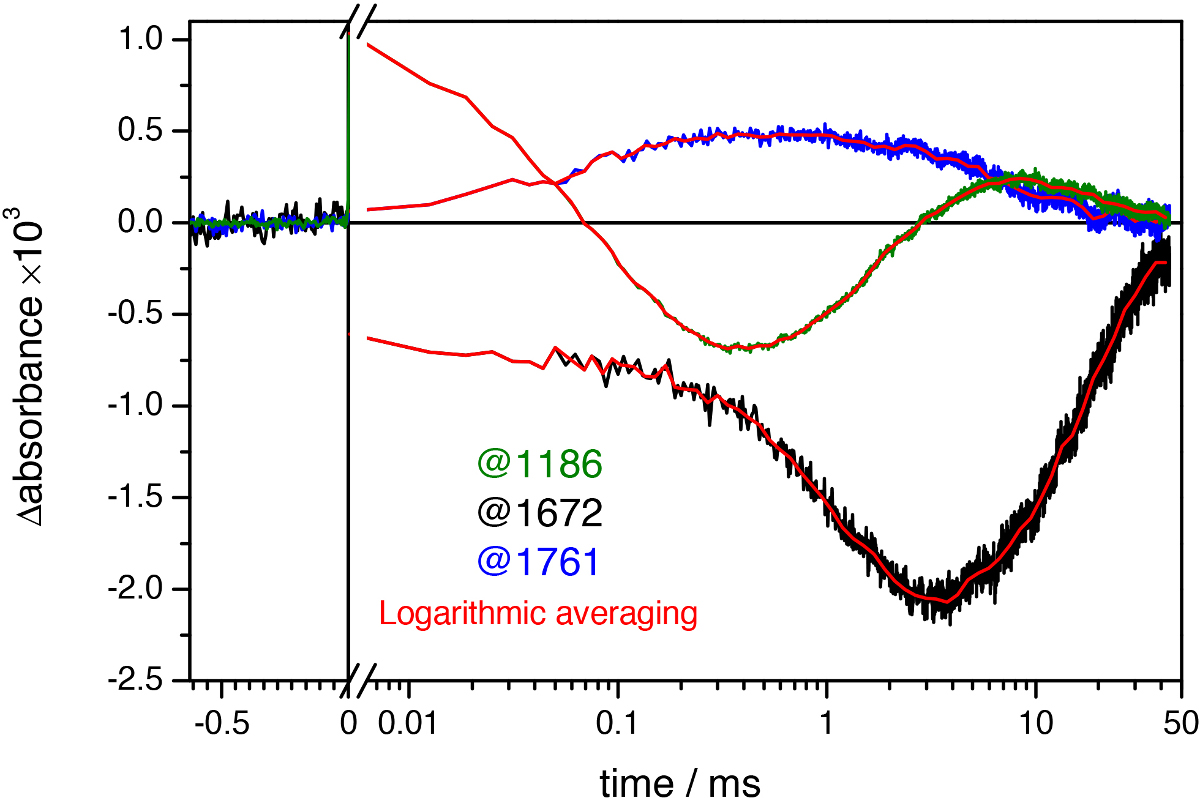
Figure 9. Light-induced absorbance changes of bR at three representative wavenumbers at uniform time spacing intervals (green, black, and blue lines) and after quasi-logarithmic averaging to ~20 points/decade (red lines). Notice the noise reduction after logarithmic averaging, revealing oscillation of the time traces in the millisecond range.

Figure 10. 3D representation of light-induced absorbance changes for bR recorded by ATR at pH 7.4 (4 M NaCl, 100 mM NaPi). A) Raw data. B) Data reconstructed with five SVD components. Please click here to view a larger version of this figure.
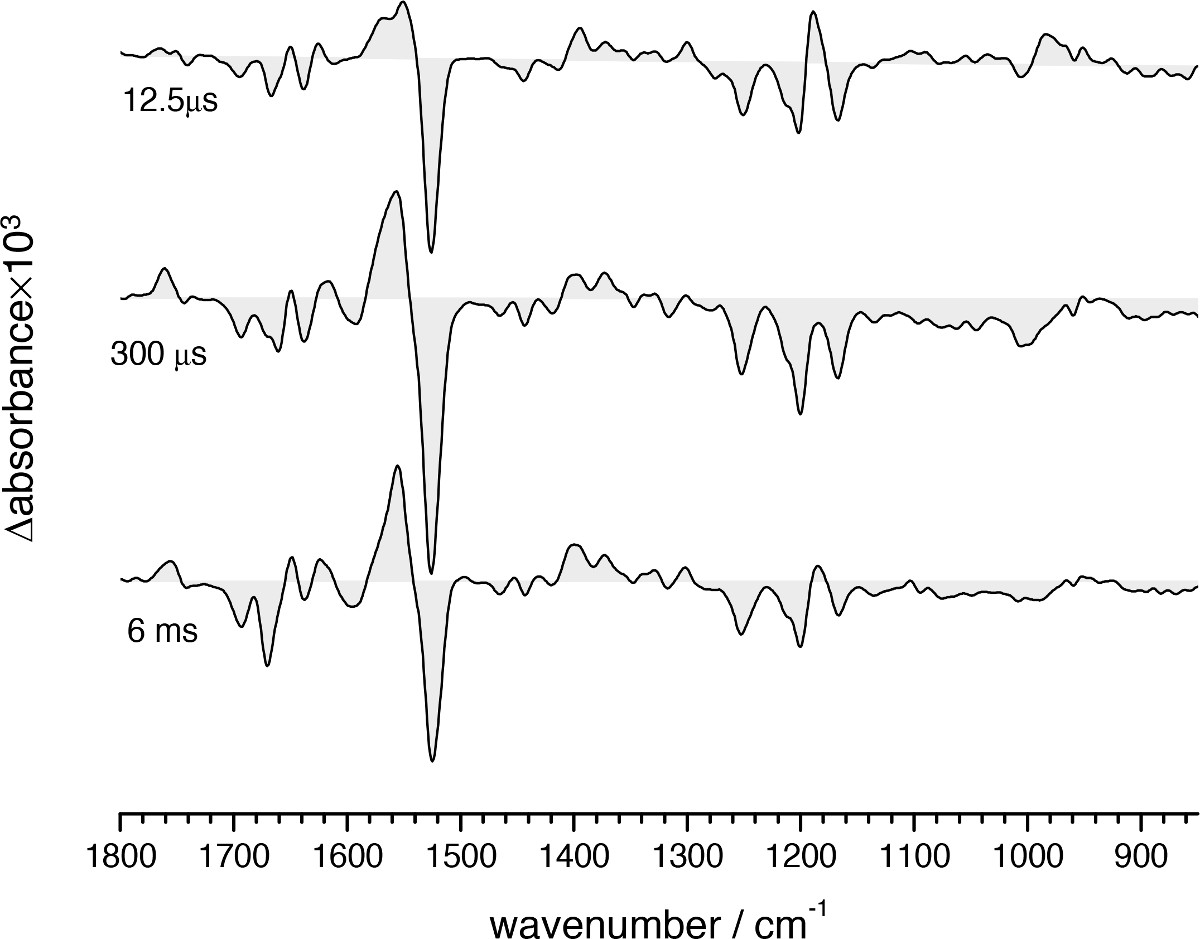
Figure 11. FT-IR difference absorption spectra of the bR photocycle at three selected times where the L (12.5 µsec), M (300 µsec), and N (6 msec) intermediates are most enriched.

Figure 12. Proton transfer and protein backbone dynamics in the bR photocycle as resolved by time-resolved step-scan FT-IR difference spectroscopy. The absorbance changes at 1,762 cm-1 reports on the protonation/deprotonation dynamics of Asp85, and at 1,741 cm-1 on the deprotonation/reprotonation dynamics of Asp96 (including H-bonding changes before 300 μsec). The time-traces at 1,670 and 1,555 cm-1 report changes in amide I and II vibrations, both sensitive to the conformation of the peptide backbone. The back traces correspond to the raw data, and the red ones to SVD-treated data. Please click here to view a larger version of this figure.

Figure 13. Singular value decomposition (SVD) of the experimental time-resolved step-scan FT-IR data of the bR photocycle (see Figure 10A). SVD was performed after weighing the experimental data taking into account the noise standard deviation dependence on wavenumber (Figure 15); combined with the 1st derivative to reduce the statistical weight of baseline fluctuations, as described before57. A) Plot of the relative singular values of the first 50 components (black circles). The first five components are assigned to signal components (red circles). The rest of components decay exponentially as expected for noise-components (see dashed gray line). B) First eight abstract spectra (U1 to U8). C) First eight abstract time-traces (V1 to V8). The abstract spectra and time-traces assigned to signal are depicted in red lines, and with black lines otherwise. Please click here to view a larger version of this figure.

Figure 14. 3D representation of light-induced IR absorbance changes for ChR2 recorded by step-scan in transmission mode. The step-scan data extends until 125 msec, only covering a part of the photocycle. A) Raw data. B) Data reconstructed with five SVD components. Please click here to view a larger version of this figure.
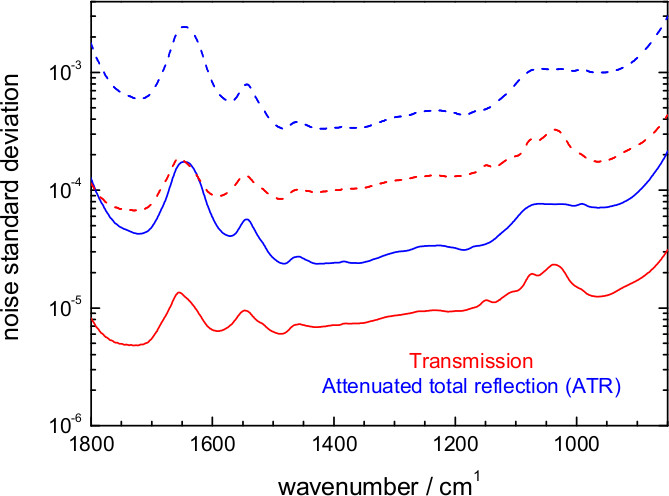
Figure 15. Estimated noise level in an FT-IR. Difference absorption spectrum at 6.25 μsec temporal and 8 cm-1 spectral resolution for an experiment involving 1 co-addition/mirror position (500 photoreactions) or ~200 co-addition/mirror position (105 photoreactions). Values are shown for the attenuated total reflection (ATR) and the transmission setup.
