Figure 1 – 4 illustrate some of the steps within the protocol. In Figures 1 and 2, the leaves around the inflorescence buds are cut to expose them to the Agrobacterium cells and the different bud stages that were used to develop the protocol. Figure 3 shows the process of flax floral-dip. Figure 4, shows an example of how the main and side branches can be labeled and how individual flowers can be tracked and identified. Figure 5 shows how the T1 progenies can be germinated on the MS plant media and then transplanted to soil for maturity. Figure 6 illustrates how wild-type flax can escape high concentrations of kanamycin, confirming previous findings in the literature6,9,14.
Figure 7 shows an example of direct PCR amplification from positive T1 transformants. The T1 flowers were collected from the main and side shoots of a single T0 plant. As can be seen from the direct PCR, 8/12 T1 plants tested positive by PCR and have amplified the different regions across the T-DNA. Our primers were also designed between the LIS-1 insert and the multiple cloning sites (Figure 7B and C). We used additional primers from the plant binary vector to amplify different segments of the T-DNA, such as the left border and the NOS terminator (data not shown) or the right border and the multiple cloning site (Figure 7D). Primers specific to the LIS-1 insert were also used in this protocol (data not shown). A list of primers is provided in Table 1. However, the sequences of these primers depends on the sequence of the T-DNA plant binary vector and the insert used for the floral-dip. We also noted that there was no significant difference in the transformation rate between flowers collected from the main and side branches.

Figure 1. Cutting the leaves around the primary inflorescence buds to expose them to the Agrobacterim cells. (A) The buds are covered by leaves. (B) Leaves have been cut around the buds to expose them. (C) Magnified image from plant in (A) after cutting to expose buds. Please click here to view a larger version of this figure.

Figure 2. The different bud stages that were used in this protocol to determine the best stage to use for the floral dip. (A) The early stage bud is approximately 2 mm. (B) The middle stage bud is approximately 5 mm. (C) The Late stage bud is approximately about 1 cm. Please click here to view a larger version of this figure.

Figure 3. The process of flax floral-dipping. (A) The primary inflorescences are dipped in the infiltration media containing the Agrobacterium cells. (B) Magnified from (A). (C) The dipped plants are laid flat until the next day, and the dipped branches are covered with plastic to maintain high humidity. Please click here to view a larger version of this figure.

Figure 4. The process of flower tracking and seed collections, from the T0 treatedplants. (A) An example of the whole plant with the main branch (the tallest branch in the center) and the side branches. (B – D) An example of the flowers from the different branches. (E) An example of the seeds collected from individual flowers (labeled a – k) from the main branch. Please click here to view a larger version of this figure.

Figure 5. The T1 seedlings are grown without antibiotic selection. (A) T1 seeds are germinated on the MS plant media. (B) Positive transformants, as determined by direct PCR, are transplanted to soil and grown to maturity. Please click here to view a larger version of this figure.

Figure 6. Antibiotic escape, a problem for T1 selection, is overcome by direct PCR screening. (A) Wild-type flax seeds germinated on MS plant media without antibiotic. (B) Wild-type flax seeds germinated on MS plant media+ increasing concentrations of kanamycin (200 µg/ml, 600 µg/ml, 1 mg/ml). (C) Wild-type flax seeds germinated on MS plant media with 2 mg/ml kanamycin. (D) PCR from wild-type flax and T1 seedling using kanamycin primers, all amplified the kanamycin gene (legends: EZ1: DNA marker, FlaxS, wild-type flaxS, C: control non-dipped branch, Ma: T1 progeny from flower “a” collected from the main branch, Sc,Sd,Se,Sf: T1 progenies of different flowers “c,d,e,f” collected from the side branch, W: no-DNA). Please click here to view a larger version of this figure.
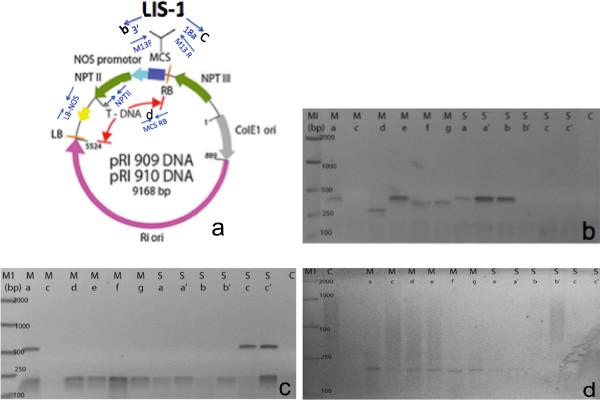
Figure 7. An example of successful PCR amplifications of T1 progenies using the direct PCR method. (A) Diagram of the Plant binary vector+ the cloned LIS-1 insert. Blue arrows indicate the position of PCR primers used in the direct PCR screening (modified from Takara). (B) PCR with primers M13F+3’ (C) PCR with primers M13R+18a (D) PCR with primers right border (RB) and multiple cloning site (MCS) **Each lane represents T1 from individual flowers collected from C: control branch (non-dipped), M: main branch flower a-g, S: side branch flower b-c’. Please click here to view a larger version of this figure.
| Forward primer sequence source | sequence 5'-3' | Reverse primer sequence source | Sequence 5'-3' | Annealing Temprature (°C) | Extension Time (Sec) | Expected size (bp) |
| M13F (T-DNA) | CTGCAAGGCG ATTAAGTTGG |
3' (LIS-1 insert) | GAGGATGGAA GATGAAGAAGG |
57 | 40 | 450 |
| 18a (LIS-1 insert) | TATTTTAACCC TATCTCCCAACAC |
M13R (T-DNA) | ATTAGGCACC CCAGGCTTTA |
57 | 40 | 520 |
| MCS (T-DNA) | TGGTCATAGC TGTTTCCTGTG |
RB (T-DNA) | TTTAAACTGA AGGCGGGAAA |
60 | 20 | 200 |
| LB (T-DNA) | TTTGATGGTG GTTCCGAAAT |
NOS (T-DNA) | GAATCCTGTT GCCGGTCTT |
60 | 30 | 380 |
| NPTII (T-DNA) | GCGATACCGT AAAGCACGAG |
NTPII (T-DNA) | GCTCGACGTT GTCACTGAAG |
65 | 45 | 502 |
Table 1. Some of the primers used for the direct PCR testing.
