Efficient Generation of hiPSC Neural Lineage Specific Knockin Reporters Using the CRISPR/Cas9 and Cas9 Double Nickase System
Summary
Genome editing tools such as the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) system have greatly improved gene targeting efficiency in human induced pluripotent stem cells (hiPSCs). This manuscript describes a protocol for generating lineage specific hiPSC reporter using CRISPR/Cas system assisted homologous recombination.
Abstract
Gene targeting is a critical approach for characterizing gene functions in modern biomedical research. However, the efficiency of gene targeting in human cells has been low, which prevents the generation of human cell lines at a desired rate. The past two years have witnessed a rapid progression on improving efficiency of genetic manipulation by genome editing tools such as the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) system. This manuscript describes a protocol for generating lineage specific human induced pluripotent stem cell (hiPSC) reporters using CRISPR/Cas system assisted homologous recombination. Procedures for obtaining necessary components for making neural lineage reporter lines using the CRISPR/Cas system, focusing on construction of targeting vectors and single guide RNAs, are described. This protocol can be extended to platform establishment and mutation correction in hiPSCs.
Introduction
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) system, together with other genome editing tools, has revolutionized the human genome manipulation in recent years1-7. Major components in the CRISPR/Cas9 system are single guide RNA (sgRNA) and Cas9. Cas9 is an RNA-guided type II DNA endonuclease. It has two nuclease domains, HNH and RuvC, which are responsible for making DNA double-stranded breaks (DSBs) at a specific genomic region next to a protospacer adjacent motif (PAM)7-10.An sgRNA has a combined function of that of crRNA and tracrRNA, two adaptive immunity RNA molecules identified in bacteria (such as Streptococcus pyogenes) and archaea to fight against DNA invasion of exogenous sources9-11. When appropriately designed and co-introduced to mammalian cells, the sgRNA recognizes the PAM sequence, base-pairs with the complementary strand of target DNA, and guides and transactivates Cas9 to cleave at both DNA strands immediately upstream of PAM7. Subsequently, homologous directed recombination (HDR, in the presence of a targeting vector) or non-homologous end joining (NHEJ) will occur to repair the DSBs. Transgenes such as cDNAs, reporter cassettes, or antibiotic resistant fragments can be integrated, and transgenic or knockin lines made.
Although the CRISPR/Cas9 system is highly efficient and relatively specific, undesired off-target activities have been reported12-15. To minimize off-target events, Cas9 nickases (Cas9n) have also been engineered9,10,14,16,17. Cas9n (Cas9 D10A or Cas9 H840A) is Cas9 enzyme with mutations at one of the two nuclease domains, which only elicits a nick at one strand of DNA. To achieve cleavage at both strands, two sgRNAs are designed to guide a pair of nickases, which cuts separately at nearby loci of the two different DNA strands. The “double nicking” strategy requires a more stringent design than the conventional Cas9 platform, and offers both high efficiency and high specificity for gene editing experiments.
This manuscript describes a protocol for generating lineage specific human induced pluripotent stem cell (hiPSC) reporter using CRISPR/Cas system assisted homologous recombination. Steps for obtaining the necessary components for making neural lineage reporter lines using the CRISPR/Cas system mediated genome editing are described, with a focus on construction of targeting vectors and sgRNAs. This protocol has been successfully repeated in multiple hiPSC lines including those derived from healthy individuals and from patients with CNS diseases. Genes targeted in our laboratory include OLIG2, HB9 (MNX1), NEUROG2, SOX1, ALDH1L1 and SOD1. Targeting efficiency from CRISPR/Cas system mediated homologous recombination in hiPSCs ranges from 20-40%, which is consistent with previous reports1,18,19. Compared to a typical efficiency of 1-2% in conventional gene targeting experiments, the targeting efficiency is significantly improved.
Protocol
1. Design and Vector Construction for Targeting Vectors
- Once the lineage specific marker is determined, locate the genomic sequence of the gene and design the homology arms accordingly. The length of 5’ homology arm is ~1 kb and of 3’ homology arm is ~1.5 kb (Figure 1).
- Tag the reporter cassette sequentially downstream of the genomic sequence right before the stop codon, by subcloning, so that the endogenous expression is not altered or disrupted. Link the reporter, or dual reporter cassette(s), with self-cleaving 2A peptide sequences (F2A, E2A or T2A) 20,21, or internal ribosome entry site (IRES).
Note: Reporters may include fluorescent proteins such as GFP, tdTomato, tagRFP, YFP, luciferase, or antibiotic resistance cassette, a combination of fluorescent protein and antibiotic resistance cassette. An example is shown in Figure 1 for constructing targeting vector for human ALDH1L1 gene, tagged by EGFP and neomycin resistance cassette. - Add a positive selection cassette downstream of the reporter. The positive selection is a floxed antibiotic resistant cassette which will be excised from the targeted iPSCs after the positive clones are selected and isolated.
- Include a negative selection cassette outside of the 3’ homology arm. Common negative selection cassettes are thymidine kinase, TK, and diphtheria toxin A (DTA) 22.
- Obtain a human BAC clone containing the sequence of the lineage marker and verify the clone by PCR amplification. Construct the targeting vector in pKD46 containing-DH5α using recombineering to pull down the targeting site 23. Apply multisite gateway, Golden gate strategies23,24 when necessary.
2. Design and Vector Construction of sgRNAs for CRISPR/Cas9 System.
- Determine the lineage specific marker that will be targeted to make the reporter. Copy the cDNA sequence from NCBI database. Go to the human BLAT search to obtain genomic DNA alignment.
- Click the tab “browser” to view the assembly of the gene. Under “View” tab, click “DNA” to obtain the full genomic DNA sequence in FASTA format.
- Locate the stop codon within the DNA sequence, where the 2A sequence and the reporter cassette will be tagged.
- Search for the protospacer adjacent motif (PAM, i.e., NGG) within the vinicity (~100 bp flanking area) of the stop codon. Scan both strands to locate the appropriate NGG sequence. Consider the specificity of the upstream sequences to avoid potential off-target events. Use open source tools such as BLAST and CasOT (see Step 5 for details).
- Copy the sequence of 20 bases immediately upstream of NGG and paste it into the web designing tool ZiFit to design oligos with overhangs compatible with human-gRNA-expression vector (with U6 promoter) MLM3636.
- Synthesize both forward and reverse oligos 7.
- To make double-stranded sgRNA, denature both oligos at 70 °C for 15 min, and gradually cool down to RT. For a 50 µl reaction, add 22.5 µl of forward and reverse oligos (stock concentration is 0.1 mM) respectively, and 5 µl of annealing buffer (100 mM Tris-HCl, 10 mM EDTA, 1.5 M NaCl, pH 8.0).
- Mix and ligate the oligos obtained from step 2.7 with BsmBI-digested human-gRNA-expression vector (with U6 promoter) MLM3636. For a 20 µl reaction, add 1 µl of oligos from Step 2.7, 100 ng of MLM3636, and 1 µl (400 U) of T4 ligase. Incubate at RT O/N.
- Transform DH5α competent cells with the ligation product by heat shock at 42°C for 30 sec. Grow the bacteria in LB agar Ampicillin (100 μg/ml) plate at 37°C O/N. Pick 3~5 colonies for plasmid miniprep according to manufacturer’s protocol. Sequence the plasmid DNA to confirm using commercial vendors. Maxiprep the correct plasmid according to manufacturer’s protocol.
- Evaluate sgRNA by T7 endonuclease 1 (T7E1) assay (see Step 4 for details). Transfect hiPSC with the plasmid and sgRNA will be expressed under the built-in U6 promoter in the MLM3636 backbone (See step 6 for details).
3. Design and Construction of Cas9n Double Nickase sgRNA
- Obtain double nickase Cas9n D10A vector pX335-U6-Chimeric_BB-CBh-hSpCas9n (D10A).
- Design a pair of forward and reverse oligos for each targeting locus. Copy the sequence of 20 bases immediately upstream of NGG as described in Step 2 and paste it into web designing tool ZiFit
- To obtain oligos with overhangs compatible with pX335-U6-Chimeric_BB-CBh-hSpCas9n (D10A), remove the first and last base of both forward and reverse oligos that are shown by the ZiFit designing tool.
- Synthesize forward and reverse oligos7. Name these oligos as f(n)-F, f(n)-R for PAMs on one side of the target site, r(n)-F, r(n)-R for PAMs on the other side of the target site. Of these, n is the number to designate different PAMs, lowercase f and r represent the location of the PAMs either at sense (f) or antisense (r) strand,, F and R represent forward and reverse oligos.
- Test multiple pairs (e.g., r5 and f2) using the T7E1 assay (detailed in Step 4) to identify the optimized pair of sgRNAs to be used for Cas9n.
- To make double-stranded sgRNA, denature both oligos at 70 °C for 15 min, and gradually cool down to RT. For a 50 µl reaction, add 22.5 µl of forward and of reverse oligos (stock concentration is 0.1 mM) respectively, and 5 µl of annealing buffer (100 mM Tris-HCl, 10 mM EDTA, 1.5 M NaCl, pH 8.0).
- Mix and ligate the oligos obtained from step 3.5 with BbsI-digested pX335-U6-Chimeric_BB-CBh-hSpCas9n (D10A). For a 20 µl reaction, add 1 µl of sgRNA from Step 3.5, 100 ng of Cas9n (D10A), 1 µl (400 U) of T4 ligase. Incubate at RT O/N.
- Transform DH5α competent cells with the ligation product by heat shock at 42 °C for 30 sec. Grow the bacteria in LB agar Ampicillin (100 μg/ml) plate at 37 °C O/N. Pick 3~5 colonies for plasmid miniprep according to manufacturer’s protocol. Sequence the plasmid DNA to confirm using commercial vendors. Maxiprep the correct plasmid according to manufacturer’s protocol.
- Evaluate sgRNA by T7E1 assay (see Step 4 for details). Transfect hiPSC with the plasmid and sgRNA will be expressed under the built-in U6 promoter in the Cas9n (D10A) backbone (See step 6 for details).
4. Evaluation of sgRNAs by T7 Endonuclease1 (T7E1) Assay
- For one 24-well plate, plate 2 x 105 293FT cells per well one day prior (day -1).
- On the day of transfection (day 0), co-transfect 293FT cells with sgRNA plasmid and Cas9 expression vector. Add 0.5 µg of sgRNA vector, 0.5 µg of Cas9 expression vector JDS246 (Cas9-003), and 1.5 µl of Lipofectamine 2000.
- For negative control, add 0.5 µg of GFP expression vector (e.g., CMV promoter driven GFP in a pcDNA3 backbone) instead of sgRNA vectors.
- After 72 hr (day 3), harvest cells by adding 100 µl of DNA quick extraction buffer and triturating vigorously.
- Incubate the mixture at 68 °C for 15 min, then 95 °C for 8 min. This is the template for the subsequent PCR experiments.
- Design both forward and reverse PCR primers to amplify the sequence flanking PAM. Usually the targeting PCR product is around 400-500 bp in length. Synthesize the oligos7.
- For a 50 µl PCR reaction, add 1 µl of template from Step 4.5, 0.5 µl of Herculase II, 0.5 µl of DMSO, 10 µl of 5x buffer, 0.5 µl of dNTP mix, and 1 µl of forward and reverse primer mix.
- Use the following PCR parameters: 98 °C for 2 min, 35 cycles of 98 °C for 20 sec, 59 °C for 20 sec, 72 °C for 1 min, with a final extension at 72 °C for 5 min.
Note: The PCR parameters described here yield specific products for several genes tested. However, these conditions are dependent upon user-chosen genes of interest. PCRs using genomic DNA as template often require optimization to ensure that no extra bands appear. These extra bands can be confused with cleavage products after the T7E1 assay. - Measure DNA concentration of the PCR products using a spectrometer at A260.
- Add 400 ng of PCR products to 2 µl of Digestion buffer 2. Add ddH2O to a final reaction volume of 19 µl. Incubate in boiling water for 5 min. Let the reaction sit on the bench and naturally cool down to RT (45-60 min).
- Add 1 µl (10 units/µl) of T7E1, incubate at 37 °C for 15 min. Stop the reaction by adding 2 µl of 0.25 M EDTA mixed with 6x DNA Loading Dye.
- Dissolve 2.5 g of agarose powder to 100 ml of 1xTAE. Microwave the mixture and pour gel onto a gel box. When gel is formed, load the whole 20 µl reaction to the 2.5% agarose gel. Run the gel at 100-150 V for 30 min.
- Inspect the gel under UV light (Figure 3). T7E1 cuts at the mismatched sites. For sgRNAs that have caused non-homologous end joining, the PCR products will show additional fast migrating bands (smaller in size) compared to negative control (described in step 4.3).
- Estimate and rank sgRNA cutting efficiency and insertion and deletion percentage (indel%) by comparing the band density using Image J software.
- For each sample that run on the gel, designate the gel bands as uncut band, cut band 1 and cut band 2.
- Measure the density of each band using ImageJ according to the software handbook.
- Calculate fcut using the following formula: fcut = (cut band 1 + cut band 2) / (uncut band + cut band 1 + cut band 2)
- Calculate Indel% using the following formula: Indel=1- √(1-fcut).
5. In Silico Prediction of Potential Off-target Sites
Note: Potential off target sites can be predicted using an online open source tool called CasOT17, which is a Perl based program.
- For a Windows system, download the CasOT program and the human genome database files to local hard drive.
- Prepare the target file of the designed sgRNA in the FASTA format.
- Open the file named “casot”. A command window appears. Leave it as is for now.
- Open the file named “opt-generator” to generate option string. Paste the path and name of the target file into the dialog of “-t” or “-target” option.
- Paste the path of the genome sequence file into the dialog of “-g” or “-genome” option.
- Input values of –s or –seed (default value is 2) and –n or -nonseed (default value is 255). The option string is generated and will appear in the box at the top of the page.
- Copy the option string and paste it into the CasOT command window described in Step 5.3.
Note: It might take 30-90 min to run the CasOT program depending on the options and the computer used. Once completed, a folder named ‘<input_target_file>-<genome_file>-<off-target_related_options>’ will be generated. Two files will be included in the folder, a “.stat” file describing the statistics and a .csv (comma-separated values) file containing the detailed search results.</off-target_related_options></genome_file></input_target_file> - Open the .csv file with a spreadsheet program. The following information is provided in the file for every off target site: chromosomal and genomic location, sequence of the site, mismatch (Mm) type, total number of mismatched bases, and exon information relating to the off target site.
- Design PCR primers for the sites that have <2 Mm in either the seed or the non-seed sequence.
- Perform PCR for selected positive hiPSC clone using the following conditions: 98 °C for 2 min, 35 cycles of 98 °C for 20 sec, 59 °C for 20 sec, 72 °C for 1 min, with a final extension at 72 °C for 5 min.
Note: The PCR parameters described here yield specific products for several genes tested in our lab. However, these conditions are dependent upon user-chosen genes of interest. PCRs using genomic DNA as template often require optimization to ensure that no extra bands appear. - Sequence the PCR products, align sequencing results with predicted sequences and search for mutations. If mutations exist, then off-target events are identified.
6. Electroporation of hiPSCs to Make Neural Lineage Reporters
- Culture hiPSCs in TeSR-E8 medium on Geltrex coated dishes. Change medium to MEF-CM (Mouse embryonic fibroblasts conditioned medium) medium two days before electroporation. Incubate in 2 ml of Accutase for 5 min and collect cells.
- Electroporate hiPSCs with linearized targeting vector, Cas9-003 expression vector, and sgRNA vector using the following conditions: 1 x 107 hiPSCs, co-transfect 2.5 µg of sgRNA, 2.5 µg of Cas9 and 50 µg of targeting vector. For double nickase strategy, add 50 µg of targeting vector, 2.5 µg of each sgRNA (with Cas9n co-expressed by the vector).
- Select clones by positive and negative selections as described24,25.
- Identify positive clones by Southern blot analysis or long range genomic PCR24,25.
Representative Results
Targeting vectors with all necessary components including homology arms, reporter cassette, positive and negative selection cassettes are constructed (Figure 1). Based on the endogenous genomic locus, multiple (2 to 3) sgRNAs (if using the Cas9 system) or sgRNA pairs (if using the Cas9n double nickase strategy) are designed and constructed into human-gRNA-expression vector MLM3636 or pX335-U6-Chimeric_BB-CBh-hSpCas9n (D10A) backbones (Figure 2). Before transfecting hiPSCs, the sgRNAs are evaluated in 293FT cells by T7E1 assay (Figure 3). Off-target sites are predicted. sgRNAs with modest cleavage efficiency (indel% between 10-30%) and fewer potential off-target sites as predicted by in silico procedure (Step 5) will be used to transfect hiPSCs by electroporation. After successful transfection and selection, positive clones are identified. If needed, PCR of potential off-target sites are performed using the genomic DNA isolated from positive clones. Sequencing or T7E1 assay will be performed to determine whether there are off-target events.
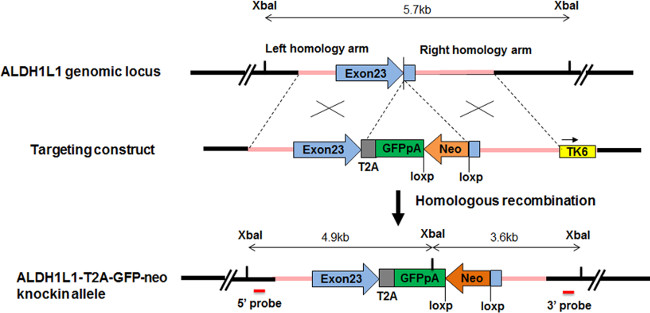
Figure 1. Design of targeting vector and sgRNA for making knockin ALDH1L1-GFP reporter. ALDH1L1 genomic sequence is tagged by GFP connected by T2A. The endogenous ALDH1L1 gene is intact and both ALDH1L1 and GFP are driven by the endogenous ALDH1L1 promoter. A floxed neomycin cassette is used for positive selection. Tk6 cassette outside of 3’ homology arm is for negative selection.

Figure 2. Two sgRNAs are designed for CRISPR/Cas9 double nicking mediated homologous recombination. PAM sequences are underlined. Stop codon is boxed. Blue Bases highlighted in blue indicate sequences for sgRNA design.
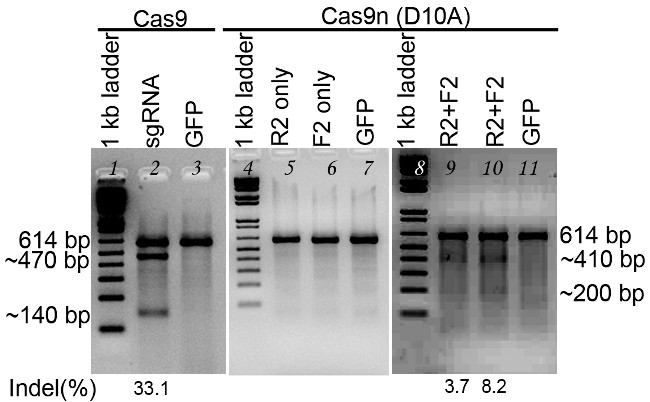
Figure 3. Evaluation of sgRNA mediated cleavage using T7E1 assay. Lanes 1~3 are transfected with Cas9 expression vector, and Lanes4~11 are transfected with Cas9n (D10A) nickase expression vector. Lanes 1,4 and 8, 1 kb DNA ladder; Lane 2, sgRNA for OLIG2 targeting using Cas9; Lanes 3, 7, and 11, GFP control; Lane 5, sgRNA binding sense strand only; Lane 6, sgRNA binding antisense strand only; Lanes 9 and 10, a pair of sgRNAs for both DNA strands. The sgRNAs are designed for targeting the human OLIG2 gene. PCR products are denatured and re-annealed. T7E1 is added to digest the heteroduplex DNA. The undigested PCR product is about 614 bp. After digestion, in the reaction for Lane 2, a portion of PCR products are digested into two bands, ~470 bp and ~140 bp, with an indel of 33%. In the reactions for Lanes 9 and 10, the two digested bands are ~410 bp and ~200 bp, with indels of 3~8%.
Discussion
Gene targeting is an essential tool in characterizing gene functions. However, the relatively low efficiency demands labor-intensive and time-consuming work. Recently development on genome editing tools such as the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) system has greatly improved the targeting efficiency. This manuscript describes a protocol for generating lineage specific human induced pluripotent stem cell (hiPSC) reporter using CRISPR/Cas system assisted homologous recombination. Steps for obtaining necessary components for making neural lineage reporter lines using the CRISPR/Cas system mediated genome editing are described, with a focus on construction of targeting vectors and single guide RNAs for both Cas9 and Cas9n double nickase system.
The CRISPR/Cas9 system can serve a variety of genetic engineering needs in hiPSCs. It can be used to make knockout and knockin (e.g., reporter) lines, inducible platform lines, as well as mutation correction. Depending on the purpose of genetic modifications, the corresponding targeting vectors may be designed and constructed, so that homologous directed recombination (HDR) other than the error-prone non-homologous end-joining (NHEJ), occurs after DSB is induced by Cas9. If the purpose is to generate missense mutations at certain locus, transfection of sgRNA and Cas9 is sufficient and targeting vectors are not necessary.
For every GOI or specific locus to be targeted, usually it would not be difficult to locate PAM (NGG) sequences, and 2 to 3 sgRNAs are therefore designed accordingly, so that the “best” or the most suitable one can be selected after being evaluated by T7E1 or SURVEYOR assay. However, because the 20-nucleotide sequence within the sgRNA that directs the cleavage of Cas9 can tolerate up to 5 mismatches16, potential off-target mutations might occur12-15. More complex designs are required for sgRNAs used in the more specific double nickase Cas9n (D10A) platform, as there are serious design constraints for using the Cas9n (D10A) with 2 sgRNAs as a 'paired nickase' system, and special attention is needed for identifying PAMs from both DNA strands. First, the distance (offset) between PAMs from both strands should be less than 100 bp with an optimal spacing of 10~30 bp 16,17. Second, nickases targeting PAMs with tail-to-tail (facing-out) orientation with appropriate spacing usually yield expected cleaving activity, while nickases designed for PAMs with head-to-head (facing-in) orientation rarely have expected cleavage as shown by T7E1 assay16. In addition, previous report has shown that although the double nickase strategy reduces, it does not completely eliminate, off-target events. Therefore, off-target sites are critical considerations to be kept in mind and in silico prediction should be performed for every sgRNA. Potential off-target sites need to be monitored for mutations once the reporter hiPSC lines are made.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Department of Neurosurgery, Memorial Hermann Foundation Staman Ogilvie Fund, the Bentsen Stroke Center at the University of Texas Health Science Center at Houston, and Mission Connect TIRR Foundation.
Materials
| Name of Material/Equipment | Vendor | Catalog no. |
| pStart-K | Addgene | 20346 |
| pWS-TK6 | Addgene | 20350 |
| pKD3 | Addgene | 45604 |
| pKD46 | The Coli Genetic Stock Center, CGSC | 7634 |
| PGK-neo-bpA sequence | Addgene | 13442 |
| Human BAC clones of target genes | https://bacpac.chori.org | |
| JDS246 (Cas9-003), Mammalian codon-optimized streptococcus pyogenes Cas9-3X Flag | Addgene | 43861 |
| MLM3636, Human-gRNA-ExpressionVector with U6 promoter | Addgene | 43860 |
| pX335-U6-Chimeric_BB-CBh-hSpCas9n(D10A) | Addgene | 42335 |
| sgRNA design, ZiFit | http://zifit.partners.org/ZiFiT/ | |
| Off target prediction tool CasOT | http://eendb.zfgenetics.org/casot/download.php | |
| Perl | http://www.perl.org/get.html | |
| NCBI | http://www.ncbi.nlm.nih.gov/ | |
| BLAT | https://genome.ucsc.edu/cgi-bin/hgBlat?command=start | |
| sgRNA Primer synthesis | Sigma | |
| One shot Top10 Electrocomp E. Coli Competent cells | Life Technologies | C4040-50 |
| AccuPrime Pfx SupperMix | Life Technologies | 12344-040 |
| Restriction enzymes | NEB and Life Technologies | |
| Zymoclean Gel DNA Recovery Kit | Zymo Researech | D4007 |
| DNA quick extraction buffer | Epicentre | QE0905T |
| Herculase II Fusion DNA Polymerases | Agilent Technologies | 600675 |
| Digestion buffer 2 | New England Biolab | |
| 6X DNA Loading Dye | Thermo Scientific | R0611 |
| TeSR-E8 Kit for hESC/hiPSC Maintenance | Stem Cell Technologies | 05940 |
| UltraPure Agarose | Life Technologies | 16500-100 |
| 50xTAE | Life Technologies | B49 |
| Essential 8 medium | Life Technologies | A1517001 |
| Dulbecco’s Phosphate Buffered Saline without Calcium and Magnesium | Life Technologies | A12856-01 |
| D-MEM/F12 with Glutamax | Life Technologies | 10565018 |
| D-MEM with Glutamax | Life Technologies | 10566040 |
| Fetal Bovine Serum-ES cell qualified | Life Technologies | 10439 |
| Knockout serum replacement | Life Technologies | 10828010 |
| 2-mercaptoethanol 1000X | Life Technologies | 21985023 |
| Non Essential Amino Acid | Life Technologies | 11140050 |
| Stempro Accutase | Life Technologies | A1110501 |
| Dispase | Life Technologies | 17105-041 |
| 0.25% Trypsin- EDTA solution | Life Technologies | 25200-056 |
| Geltrex | Life Technologies | 12760-013 |
| ROCK inhibitor Y-27632 | Millipore | SCM075 |
| SMC4 reagent | BD | 354357 |
| Neomycin resistant MEF | Millipore | PMEF-NL |
| Hygromycin resistant MEF | Millipore | PMEF-HL |
| G418 (Geneticin) | LifeTechnologies | 11811 |
| Hygromycin B | Life Technologies | 10687010 |
| FIAU (Fialuridine, 1-2-Deoxy-2-fluoro-ß-D-arabinofuranosyl-5-iodouracil | Moravek Biochemicals and Radiochemicals | M251 |
| Electroporator: Gene Pulser Xcell | Bio-Rad | |
| 0.4 cm electroporation cuvette | Bio-Rad | 165-2088 |
| DIG-High prime DNA labeling and detection starter kit II | Roche | 11585614910 |
| Hybridization denature solution | VWR | 82021-478 |
| PCR DIG probe synthesis kit | Roche | 11636090910 |
| DIG wash set | Roche | 11585762001 |
| Anti-Digoxigenin (DIG-AP) | Roche | 11093274910 |
| CSPD chemiluminescence system | Roche | 11755633001 |
| DIG wash and block buffer set | Roche | 11585762001 |
| 50X TAE buffer | Life Technologies | 24710030 |
| Blotting buffer (25 mM Tris pH 7.4, 0.15 M NaCl, 0.1% Tween20) | ||
| Hoefer Ultraviolet Crosslinker | Fisher Scientific | 03-500-308 |
| Spermidine | Fisher | AC13274-0010 |
| Tris HCl 2M ( pH 7.5 ) | VWR | 200064-506 |
| Denville Scientific blue bio film 8×10 | Fisher | nc9550782 |
| DNA molecular weight marker II ( DIG-labeled ) | Roche | 11218590910 |
| Amersham Blotting membrane Hybond-N+ | Roche | 95038-400 |
| Pyrex glass drying tray | Fisher | 15-242A |
| Kimberly-Clark C-fold paper towels | Fisher | 06-666-32B |
| Whatman 3MM paper ( 26X41) | Fisher | 05-713-336 |
| Hybridization bag | Roche | 11666649001 |
| Hybridization tubes | Fisher | 13-247-300 |
| Hybridization oven rotisserie Shake 'n' Stack | Fisher | HBMSOV14110 |
References
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153, 910-918 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339, 823-826 (2013).
- Hwang, W. Y., et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature. 31, 227-229 (2013).
- Friedland, A. E., et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. , (2013).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336-4343 (2013).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819-823 (2013).
- Anders, C., Niewoehner, O., Duerst, A., Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 513, 569-573 (2014).
- Deltcheva, E., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471, 602-607 (2011).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America. 109, E2579-E2586 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-821 (2012).
- Jinek, M., et al. RNA-programmed genome editing in human cells. eLife. 2, e00471 (2013).
- Fu, Y., et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology. 31, 822-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31, 827-832 (2013).
- Mali, P., et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology. 31, 833-838 (2013).
- Pattanayak, V., et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nature Biotechnology. 31, 839-843 (2013).
- Shen, B., et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature Methods. 11, 399-402 (2014).
- Ran, F. A., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 154, 1380-1389 (2013).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature Protocols. 9, 1956-1968 (2014).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154, 1370-1379 (2013).
- Szymczak, A. L., et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nature Biotechnology. 22, 589-594 (2004).
- Carey, B. W., et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proceedings of the National Academy of Sciences of the United States of America. 106, 157-162 (2009).
- Yagi, T., et al. A novel negative selection for homologous recombinants using diphtheria toxin A fragment gene. Analytical Biochemistry. 214, 77-86 (1993).
- Wu, S., Ying, G., Wu, Q., Capecchi, M. R. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nature Protocols. 3, 1056-1076 (2008).
- Xue, H., et al. A targeted neuroglial reporter line generated by homologous recombination in human embryonic stem cells. Stem Cells. 27, 1836-1846 (2009).
- Liu, Y., Jiang, P., Deng, W. OLIG gene targeting in human pluripotent stem cells for motor neuron and oligodendrocyte differentiation. Nature Protocols. 6, 640-655 (2011).
