1. In Silico Computer Modeling
- Download a PDB structure of the protein of interest from the protein data bank (PDB). Prepare the structure by removing excess subunits and then adding missing hydrogen atoms and explicit solvent using the "Cell Neutralization and pKa prediction" experiment in YASARA16. For membrane bound triterpene cyclases, suitable dimensions of the water box are 91x67x77 Å. Adjust the protonation state of the catalytically active amino acids manually.
Note: If necessary, a substrate analog in the PDB file can be modified to a real substrate by using the "Edit|Swap|Atom" command. - Minimize the structure by the "Energy Minimization" command using the AMBER force field suite17. Use the particle mesh Ewald approach (PME)18 to account for long-range electrostatic interactions. Set the cut-off for van der Waals interactions according to standard settings.
Note: The "Energy Minimization" command removes conformational stress by a short steepest descent minimization, then a simulated annealing (timestep 2 fsec, atom velocities scaled down by 0.9 every 10th step) is performed until convergence is reached (i.e., improvement of energy by less than 0.012 kcal/mol per atom during 200 steps).
2. Model of Active Site Solvation and Water Access
- After the simulated annealing, run a 20 nsec molecular dynamics (MD) trajectory and generate at least 10 snapshots by the "File|Save As|Simulation Snapshot" command followed by "Simulation On". Run the simulations on a standard computer with periodic boundary conditions under the canonical ensemble. Maintain the temperature at 298 K using a Berendsen thermostat.
- Prepare the snapshots obtained from 2.1 by deleting all water molecules and eventual ligands/substrates using the "Edit|Delete|Residue" command. Superpose each snapshot individually on the original PDB-structure by the "Superpose|Object" command to ensure that all structures share the same spatial position. Save each snapshot prepared in this way as a new PDB-file (using the "File|Save as|PDB" command).
Note: For experienced modelers: Write a script to automate this process according to the template given in Supplementary Code File 1. - Use the prepared snapshots (PDB) from 2.2 that have been aligned in 3D-space as inputs to the software CAVER 3.019. For the analysis of a limited number of snapshots, run CAVER on a standard computer. Use a probe radius of 0.7 Å19 to assure the detection of tunnels that are specific for the transport of water molecules.
- Visualize the tunnels generated by CAVER 3.0 in a molecular graphics software. For easy visualization of tunnels, use the macro shown in Supplementary Code File 2.
3. In Silico Enzyme Engineering to Modify Water Patterns and Water Dynamics
- Identify the highest ranked tunnels according to the header "ID" listed in the output file "summary.txt" generated from the CAVER 3.0 software.
- Identify amino acids lining the highest ranked tunnels (3.1) and that display a small side chain pointing towards the channel interior by visual inspection (using the model obtained from 2.4). Identify single amino acid substitutions that will block the tunnel by increased steric hindrance using the "Swap Residue" command. Verify that the tunnel is obstructed by visual inspection, and then by repeating steps 1.2-3.1 of the protocol.
4. Expression of Mutated Genes
- Introduce mutations in the wild-type gene by Mutagenesis PCR using non-overlapping primers20.
- Add plasmid DNA containing the gene of interest to tubes with competent cells of a suitable cloning strain and incubate on ice for 30 min. Perform a heat shock transformation for 45 sec in a water bath with the temperature set to 42 °C. Add 500 µl of a fresh rich medium (2YT, 16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl) and incubate at 37 °C, 200 rpm for 45 min. Make a selection by plating the transformed cells on agar plates supplemented with appropriate antibiotics.
- After verifying the DNA sequence of the mutated gene, transform the plasmid DNA into an expression strain using heat shock as described above. For the expression of membrane bound triterpene cyclases, use BL21(DE3).
- Start a 5 ml O/N culture at 37 °C of the expression strain in 2YT-media supplemented with the appropriate antibiotics.
- Inoculate a 2 L baffled shake flask, containing 300 ml 2YT-media supplemented with appropriate antibiotics, to a final OD of 0.05-0.09 (measured at 600 nm). After induction at an OD of 0.6-0.8 and protein expression, freeze the cell pellet and store at -80 °C.
- For membrane-bound triterpene cyclases in a pD861 vector, induce with 0.5 to 2 mM rhamnose and 4 hr of expression at 37 °C to achieve high yields of protein21.
5. Membrane Extraction Using a Normal Centrifuge and Protein Purification
- Prepare the following buffers: Resuspension buffer (100 mM potassium phosphate, pH 7.5 supplemented with Protease Inhibitor Tablets), Detergent buffer (1% (v/v) Triton X-100, 50 mM potassium phosphate, pH 7.5) and Reaction buffer (0.2% Triton X-100, 50 mM potassium phosphate, pH 7.5).
- For squalene-hopene cyclases or other membrane-bound enzymes with a lower pH optimum, replace the potassium phosphate in the resuspension buffer with citrate. For such enzymes use the following buffers: Detergent buffer (1% Triton X-100, 60 mM citrate, pH 6.0) and Reaction buffer (0.2% Triton-X 100, 60 mM citrate, pH 6.0).
Note: If a His-tag is used for purification, supplement the resuspension buffer with 20 mM imidazole (final concentration).
- For squalene-hopene cyclases or other membrane-bound enzymes with a lower pH optimum, replace the potassium phosphate in the resuspension buffer with citrate. For such enzymes use the following buffers: Detergent buffer (1% Triton X-100, 60 mM citrate, pH 6.0) and Reaction buffer (0.2% Triton-X 100, 60 mM citrate, pH 6.0).
- Weigh a frozen cell pellet in a glass beaker. Resuspend the cells in resuspension buffer using a homogenizer to a final concentration of 0.3 g cells/ml. Lyse the resuspended cells by ultrasonication with an amplitude of 80% and a pulse of 1 sec on and 1 sec off. Use three repeating cycles of 50 sec each.
- Add detergent buffer to a final concentration of 0.2% (v/v) detergent. Equilibrate by end-over-end mixing at 4 °C for 1 hr. Recover the membrane protein fraction by saving the supernatant after a single centrifugation step at 39,000 x g for 50 min at 4 °C.
Note: If His-tag purification is used, add approximately 1.5 ml Ni-NTA agarose/g cells and incubate for 1 hr. - Perform a first purification step by either ion exchange or His-tag purification21. Supplement the equilibration and elution buffers with 0.2% Triton X-100. Concentrate the purified protein five times using Centrifugal Filter Units with a cut-off of 10 kDa.
Note: The size of the Triton X-100 micelles results in a concentration of detergent to a final concentration of approximately 1%. - Finalize purification by a gel filtration polishing step using the concentrated protein and a matrix compatible with a fractionation range for globular proteins of 2 × 104-8 × 106. Use the reaction buffer as running buffer. Measure the amount of purified protein by using the Bradford Ultra kit according to the manufacturer's protocol.
6. Kinetics
- Make a fresh 5 mM stock solution of substrate in reaction buffer. Obtain a homogenous emulsion by ultrasonication at 30% amplitude for 2-5 min.
- Equilibrate a thermomixer at the desired reaction temperature. Verify the temperature inside a glass vial containing water by an external thermometer.
- For single-substrate kinetics, dilute the emulsified substrate into at least five glass vials to the same final substrate concentration.
- For one-pot relative kinetics with multiple substrates, prepare at least five identical glass vials by mixing all substrates in each vial. For hydrophobic triterpenes, use final substrate concentrations in the range of 10-200 µM.
- Preincubate the glass vials in the thermomixer for 10 min. Start the reaction by adding the enzyme. A total reaction volume of 1 ml is suitable. Use a shaking speed of at least 1,200 rpm.
- Stop the reactions at different time points by adding 500 µl ethylacetate spiked with 100 µM decanol as an integration standard.
7. Extraction and Thermodynamic Analysis
- Extract the reaction mixtures by vortexing and manually shaking the glass vials vigorously for 1 min. Centrifuge the glass vials at 9,600 x g in a table top centrifuge for 10 min at RT. Transfer the top layer (organic phase) to an empty tube. Add an additional 500 µl of extraction solvent to the reaction tubes and repeat the extraction procedure.
- Dry the extracted reaction mixtures with Na2SO4. Vortex for 5 sec and let the tubes rest for 10 min. Perform a final centrifugation step at 9,600 x g for 1 min at RT using a table top centrifuge. Transfer the extracted samples to gas chromatography (GC) vials.
- Repeat the steps under 6.1-7.2 for each enzyme variant of interest, using at least four different initial concentration of the substrate ( for single substrate kinetics) and four different temperatures (for both single substrate and competitive kinetics).
- Perform GC-analysis as previously described3. Use a non-polar column for the analysis of hydrophobic pentacyclic products. Set the initial oven temperature to 120 °C and use a temperature gradient of 5 °C/min, depending on the products and the column.
- Integrate the peak areas of the product(s) and transform product peak areas into the corresponding product concentrations by utilizing the area of the integration standard (corresponding to 100 µM). Plot the concentration of each product ([P]) versus the reaction time (t). Perform linear regression of the data points corresponding to less than 10% conversion. The initial reaction speed (V0) is given by the slope of the fitted line according to:
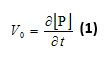
Note: For one-pot relative kinetics with multiple substrates, V0 is the initial rate of a particular substrate under the influence of other substrates. - For single substrate kinetics, plot V0/[E] against the corresponding substrate concentrations. The apparent kcat/KM value is given by the slope of the linear part of the graph according to:
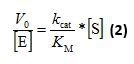
[S] is the concentration of substrate and [E] the concentration of enzyme used in the transformation. kcat/KM equates to the catalytic constant over the Michaelis constant. Repeat the analysis for each temperature and each enzyme variant. - For single substrate kinetics, perform a thermodynamic analysis according to transition state theory22
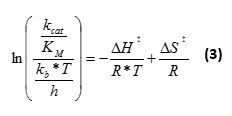
kb is the Boltzmann constant, h the Planck's constant, T the temperature in Kelvin, and R the gas constant. Perform a linear regression analysis of the plot of ln((kcat/KM)/((kb*T)/h))) versus 1/T. The activation enthalpy (ΔH‡) is given by (-slope)*R and the activation entropy (ΔS‡) is given by (intercept)*R.
Note: Likewise, an analysis under saturating substrate concentration can be performed by using kcat as rate constant in equation 3. - For one-pot relative kinetics with multiple substrates, obtain the relative apparent
kcat/KM values from23:
(kcat/KM)A/(kcat/KM)B = V0,A/V0,B*[B]/[A] (4)
for which V0,A refers to the initial rate for substrate A in competition with substrate B. - For one-pot relative kinetics with multiple substrates, obtain the relative thermodynamic parameters of activation for B, compared to A as reference, by nonlinear regression:
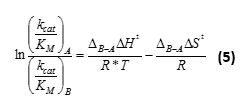
- Calculate the absolute activation parameters for substrate B from the activation enthalpy and entropy of reference compound A:
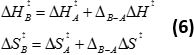
The importance of water dynamics in enzymatic polycyclization catalysis is implicated by in silico analysis and subsequent visualization of detected tunnels (using the script in Supplementary Code File 2). Following section 3 of the protocol, S168 is found to be a "hot spot" amino acid residue lining one of the tunnels in the triterpene cyclase enzyme from Alicyclobacillus acidocaldarius (Figure 1, middle left). By introducing the mutation S168F in silico, the tunnel is obstructed as seen by visual inspection (Figure 1, bottom left).
The reaction rate for the formation of polycyclic products displayed by the triterpene cyclase enzyme is highly sensitive to temperature (Figure 2A). Following the protocol (section 4-7), it is found that the apparent kcat/KM displayed by wild-type enzyme for the formation of pentacyclic products increases fiftyfold when the temperature is raised by 25 °C (Figure 2A). Blocking individual tunnels by introducing a single point mutation has a significant effect on the experimentally determined absolute apparent kcat/KM values, as well as on their temperature dependence (Figure 2A).
From the experimentally determined kinetic parameters, linear thermodynamical plots are generated by equation 3 and by following section 6-7 of the protocol for single substrate kinetics (Figure 2B). The very large changes in activation entropy and enthalpy observed for variants harboring blocked tunnels implies a key role for water dynamics in promoting the polycyclization cascade (Figure 2C, calculations based on Figure 2B). Moreover, variants with altered water tunnels display an altered Gibbs free energy of activation (ΔG‡=ΔH‡–T*ΔS‡, Figure 2B-C). For instance, at 303 K the S168F tunnel variant displays a Gibbs free energy of activation of 14 kcal/mol compared to 16 kcal/mol for the wild-type enzyme.
Following the protocol in section 7, equation 4 allows for the calculation of apparent kcat/KM values for additional substrates from one-pot kinetic experiments (Figure 3A). Moreover, a linear thermodynamic plot (Figure 3B) can be constructed by running the competition essay at different temperatures (equation 5). In analogy to single substrate kinetics, the relative activation enthalpy and entropy for additional substrates compared to a reference compound are readily accessible from the intercept and slope of the linear fits to the experimental data. Absolute values of thermodynamic parameters of activation can be assessed from arithmetic addition by using thermodynamic data associated with the reference compound (equation 6). Using the protocol herein, generated results clearly show that membrane enzyme variants with blocked water tunnels display fundamentally different thermodynamic parameters of activation for substrates of different sizes (Figure 3C).
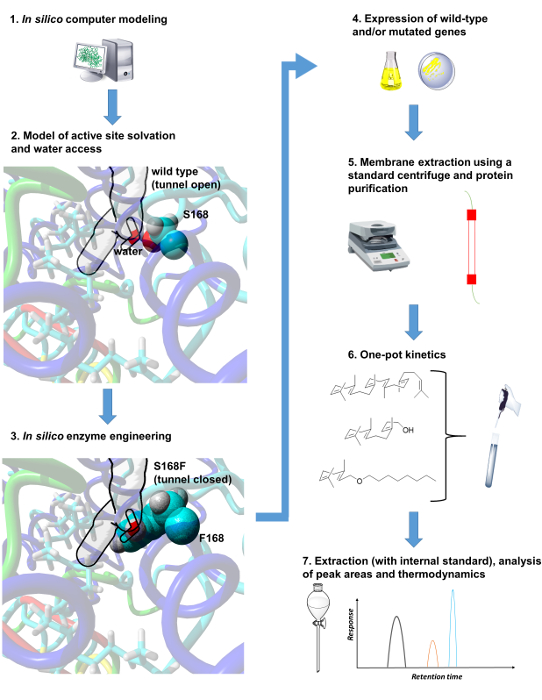
Figure 1. Protocol summary: in silico computer modeling in concert with experimental protein design and thermodynamic analysis allows for an enhanced understanding of the influence of solvent dynamics on enzyme catalysis. Please click here to view a larger version of this figure.
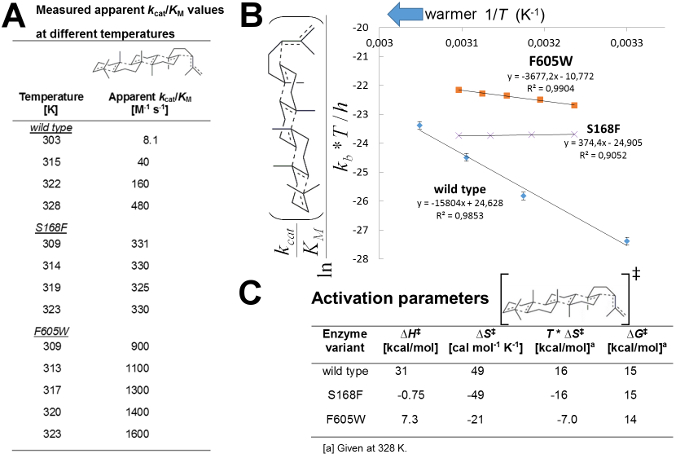
Figure 2. Thermodynamic parameters of activation of wild-type and tunnel variants from single substrate kinetics. (A) Apparent kcat/KM values obtained from experimental data and by using equations 1 and 2. (B) Thermodynamical analysis using transition state theory and linear fit of the experimental data to equation 3. (C) Thermodynamic parameters of activation calculated from the plots in (B). The prefolded squalene substrate is shown with bonds formed/broken as dotted lines. Please click here to view a larger version of this figure.
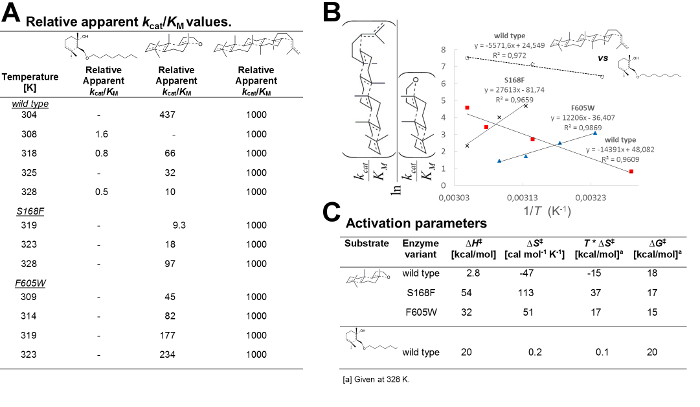
Figure 3. Thermodynamic parameters of activation of wild-type and tunnel variants for different substrates. (A) Relative kcat/KM values obtained from one-pot kinetics by using equation 4. (B) Thermodynamical plots of the kinetic data at different temperatures using equation 5. (C) Absolute thermodynamic parameters of activation calculated by equation 6 and by using the substrate squalene in Figure 2 as reference. Bonds formed/broken in the substrates are shown as dotted lines. Please click here to view a larger version of this figure.
