Consistent with previously published studies15, successful parabiotic fusion of zebrafish embryos depends on the staging and orientation of the two embryos and the concentration of methylcellulose. With just a few simple, inexpensive tools, surgically fused developing blastulae were generated that grew into parabiotic embryos with shared circulation. These tools included a modified Pasteur pipette, a 10 ml pipette pump, and wood handled teasing needles which were used either alone or with a gel-loading tip or glass microinjection needle fixed to the end by lab-film (Figure 1).
After placing the two blastulae in 4% methyl cellulose and using the gel-loading tip to orient them with their animal poles facing one another (Figure 2A), the embryos were carefully wounded at their point of contact with the glass microinjection needle (Figure 2B). A greater degree of wounding (compare Figure 2B 到 Figure 2C), and revisiting embryo pairs to bolster an initial connection with additional wounding, increased the likelihood of the embryos maintaining a connection that resulted in successful fusion (Figure 3A-C, Movie 1), and without additional morphological defects or delayed development. When done carefully, nearly 100% of the embryo pairs were fused, although a fraction of these embryos (around 25%) never established healthy circulation in both halves. By orienting the two blastulae with their animal poles directly aligned, reliable head-to-head or yolk sac-to-yolk sac fusions were generated with shared circulation. In most instances, the embryos had two hearts pumping a shared common circulation (Figure 3D).
To confirm that the embryos indeed shared their circulation, fluorescent dextran was injected into circulation, which could be seen subsequently circulating through both embryos (Movie 2). Additionally, transgenic embryos that had GFP+ erythrocytes (lcr:eGFP) were fused to embryos that had mCherry+ vascular endothelial cells (flk1:HRAS-mCherry). By 48 hpf, GFP+ erythrocytes were observed circulating through the flk1:HRAS-mCherry partner embryo (Figure 4A and Movie 3). To expand the utility of the parabiotic system, temporal control of gene expression was added. Prior to fusion, one of the two embryos was injected with an hsp70:eGFP DNA construct 20. While the fused embryos were growing at 28.5 °C, no fluorescence was observed. In contrast, after a brief 30 min heat shock at 37 °C, a clear GFP signal was visible in one of the two embryos (Figure 4B). In some instances GFP+ cells were observed circulating in the un-injected partner embryo. Thus, placing a gene of interest under the control of a heat shock promoter provides additional temporal control to studies investigating cell-intrinsic or cell-extrinsic functions of candidate genes.

Figure 1. Tools for Generating Parabiotic Zebrafish Embryos.
(A) Annotated image shows tools used for surgical fusion of developing blastulae. Tools include a modified Pasteur pipette (top), which is used in conjunction with a 10 ml pipette pump (green). Wood handled teasing needles are used either alone or with a plastic gel-loading pipette tip or pulled glass microinjection needled fixed to the end with lab-film. (B) Image shows the ends of three glass microinjection needles that have been broken with a forceps at different diameters. The needles are lying on top of a micrometer; the smallest lines are spaced 10 µm apart. Please click here to view a larger version of this figure.
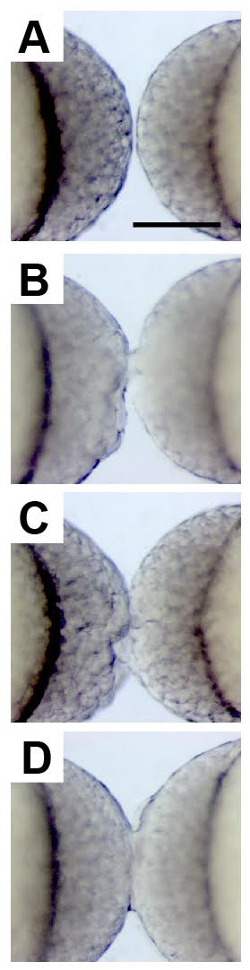
Figure 2. Surgical Fusion of Developing Blastulae.
High-magnification images show two zebrafish embryos at the high stage of development, oriented with their animal poles facing one another, prior to surgical stitching (A), immediately after minimal wounding (B), and after more substantial wounding (C). 20 min later it is apparent that the embryos have been successfully fused based on the uninterrupted bridge of cells between the two blastulae (D). Scale bar represents 250 µm. Please click here to view a larger version of this figure.
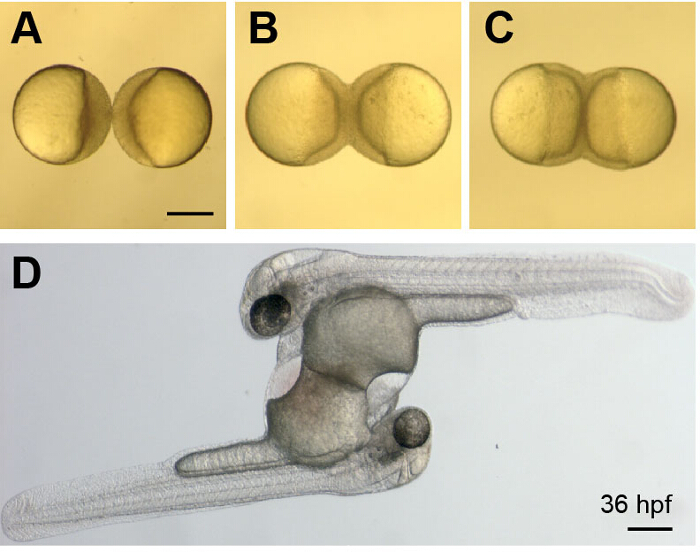
Figure 3. Development of Parabiotic Zebrafish Embryos.
(A-C) Images show a whole-parabiont view of early development just after wounding (A) and as the embryos continue to develop and undergo epiboly (B and C) These images correspond to Movie 1. (D) Image shows a parabiotic embryo pair at 36 hpf. The embryos are connected at their yolk sacs, have two hearts and share a common circulation. Scale bars represent 250 µm. Please click here to view a larger version of this figure.
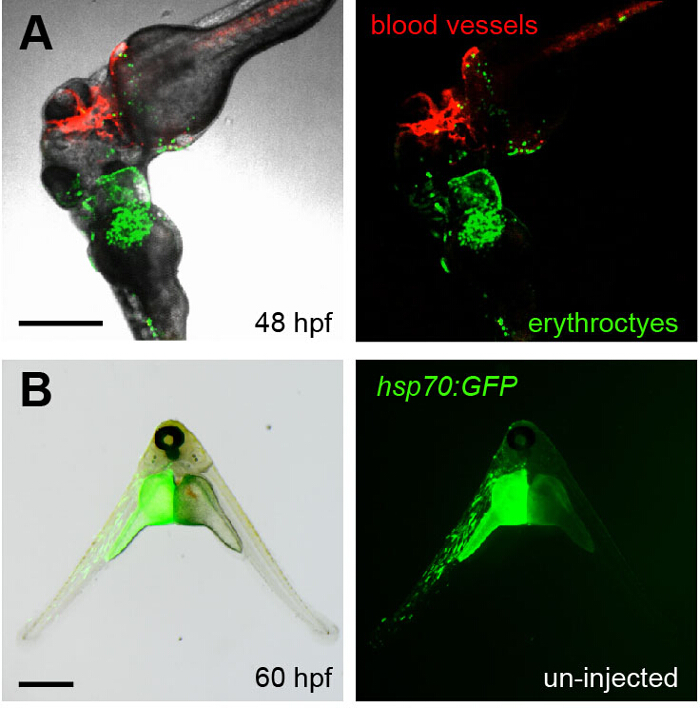
Figure 4. Visualizing Parabiosis with Genetically Encoded Fluorescent Proteins.
(A) Images (fluorescence alone, right; overlay with transmitted light, left) show the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes can be seen circulating through the partner embryo. This image corresponds to Movie 3. (B) The left embryo in this pair was injected with an hsp70:eGFP construct at the one-cell stage and then fused to an un-injected partner (right). When the animals were heat shocked at 37 °C for 30 min at 36 hpf, the green fluorescence of GFP was observed in the left embryo at 60 hpf. Scale bars represent 500 µm. Please click here to view a larger version of this figure.
Movie 1. Early development of surgically fused zebrafish blastulae. (Right click to download).
Movie shows the early development of a pair of surgically fused zebrafish blastulae. Epiboly can be seen occurring simultaneously in each embryo. This movie is related to Figure 3A-C.
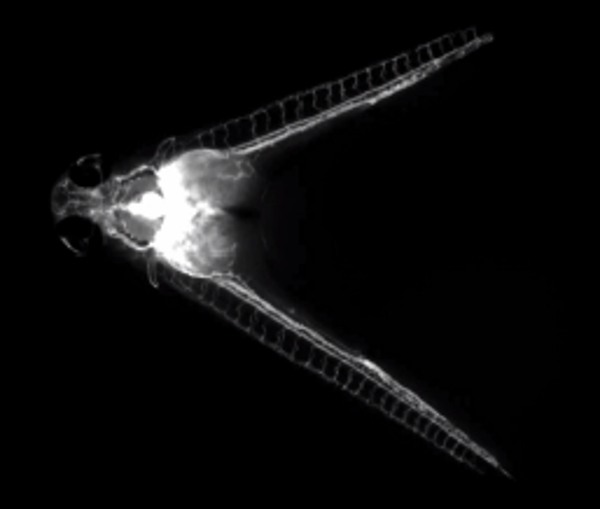
Movie 2. Fluorescent dextran injection illuminates a shared circulation. (Right click to download).
Movie shows fluorescent dextran being injected into the common cardinal vein of a parabiotic embryo pair at 60 hpf. The fluorescent dye can be seen subsequently circulating through both embryos.
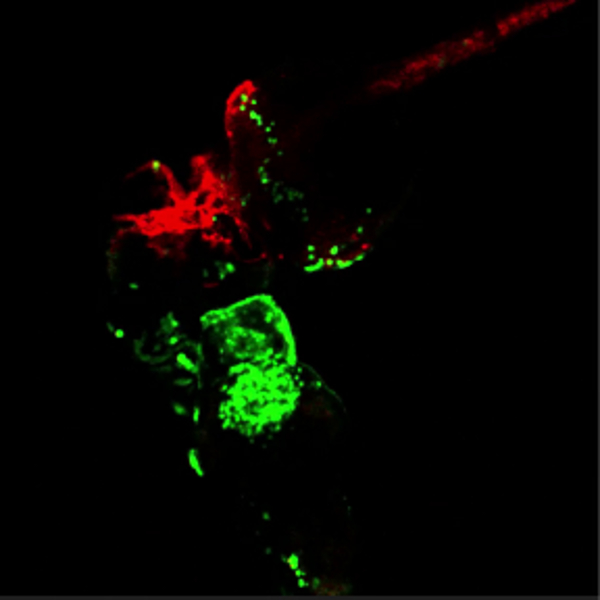
Movie 3. Green blood flowing through red vessels. (Right click to download).
Movie shows the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes are seen circulating through the partner embryo. This movie is related to Figure 4A.
