A conceptual overview of WTB-PCR during extension is presented in Figure 1. Because a single nucleotide mismatch in the blocker-DNA hybrid greatly decreases its melting temperature (ΔTm=20 – 30 °C), amplification of the WT allele is blocked while mutant template DNA is free to complete extension17. In this manner, mutant DNA is amplified exponentially while WT DNA is amplified linearly.
A demonstration of the mutant enrichment achieved by WTB-PCR is presented in Figure 2. Genomic DNA from patients with and without mutations were tested by both conventional and WTB-PCR and then sequenced to demonstrate the typical enrichment achievable by WTB-PCR and a lack of false positives in WT DNA. The working concentration of blocker used in WTB-PCR MMX should be determined by titration experiments and should achieve the desired level of mutant enrichment while not resulting in false positives or blocking amplification of WT DNA entirely. Sequencing analysis of WTB-PCR product demonstrates enrichment for the mutant allele and a limit of detection in excess of 0.5% mutant allele in a background of WT compared with 16% in conventional PCR3.
The effects of this increase in sensitivity in clinical testing may vary depending on the relative quantity of neoplastic cells in the samples tested. In a methods comparison study, the WTB-PCR assay described here has demonstrated that 64% of MYD88 mutations would be missed by conventional testing of patients with WM or MGUS3.
A characteristic drop-off in signal intensity (Figure 3) is often seen if too high a concentration of blocker is used or if post-PCR purification failed to remove blocker prior to bi-directional sequencing. The latter is demonstrated when magnetic bead purification is substituted for enzymatic purification. Though enzymatic purification is an attractive option when working with greater sample numbers, it is inappropriate for application with WTB-PCR as it fails to remove blocker from solution prior to sequencing.
An example of sequence artifacts frequently found in FFPE-derived DNA as a result of cytosine deamination (C:G>T:A) are presented in Figure 43,18,19,20. Though the actual causes of cytosine deamination are poorly understood, any PCR based assay that enriches for mutant alleles will detect these low frequency artifacts21,22. False positives due to deamination are best avoided by starting with high quality template material; in the many cases where this is not possible, treatment with uracil DNA glycosylase (UDG) during extraction can aid in limiting the frequency and intensity of deamination artifacts. UDG treatment of FFPE tissue during extraction (as part of the DNA FFPE kit) excises deaminated cytosine residues thereby preventing artificially induced C:G>T:A mutations. However, 5-methylcytosine residues that frequently occur at CpG dinucleotides are deaminated to thymine, which cannot be excised by UDG. The resulting sequencing artifacts are fairly recognizable and often appear as tandem mutations as seen in Figure 4. If samples have already been extracted, UDG treatment can be implemented in a secondary extraction with relatively low DNA loss.
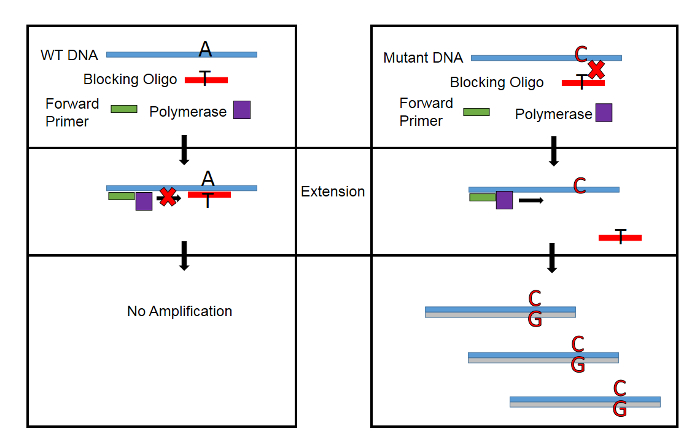
Figure 1: Conceptual Overview of WTB-PCR. A single nucleotide mismatch in the Blocker-DNA hybrid decreases Tm by up to 30 °C. By designing the blocking oligonucleotide to have a Tm of 10 – 15 °C above the temperature during extension, amplification of WT DNA is blocked while allowing amplification of mutant DNA. Please click here to view a larger version of this figure.
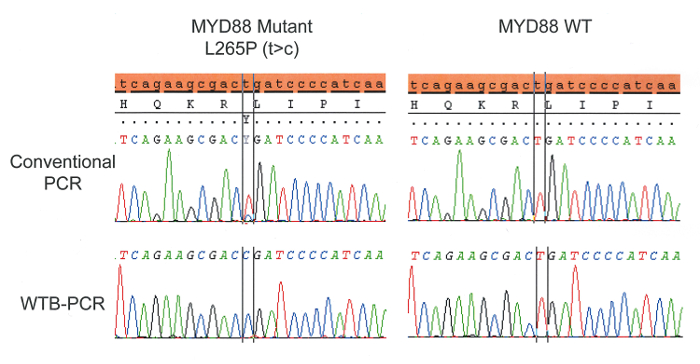
Figure 2: Genomic DNA from patients with and without mutations were tested by both conventional and WTB-PCR and then sequenced to demonstrate the typical enrichment achievable by WTB-PCR. The final concentration of blocker used to achieve WTB-PCR was selected to achieve maximum mutant enrichment while not causing false positives in WT DNA or blocking amplification of WT DNA entirely. Please click here to view a larger version of this figure.
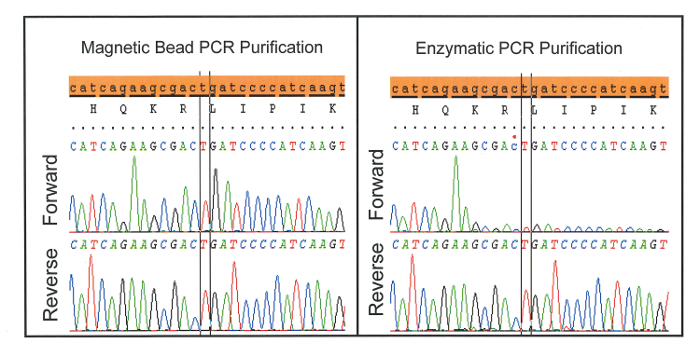
Figure 3: Characteristic drop-off in signal intensity seen when enzymatic PCR purification is used instead of magnetic beads. This is likely because enzymatic purification fails to remove blocker prior to bi-directional sequencing. Please click here to view a larger version of this figure.
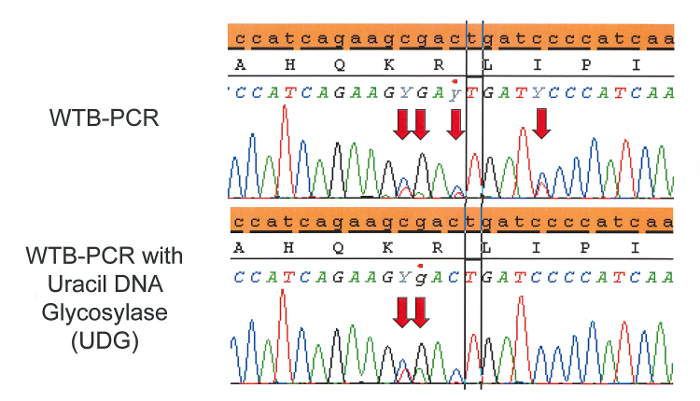
Figure 4: C:G>T:A sequencing artifacts arise in FFPE tissue when cytosine or methylated cytosine are deaminated via formalin fixation to uracil or thymine, respectively. Uracil DNA glycosylase (UDG) can excise uracil prior to WTB-PCR helping to reduce sequencing artifacts. However, thymine resulting from deaminated 5-methylcytosine, which frequently occurs at CpG islands, cannot be excised by UDG. Decreasing the concentration of blocker used in WTB-PCR may help to reduce the occurrence of sequencing artifacts that are not remedied by UDG treatment. Please click here to view a larger version of this figure.
