The assay has been benchmarked using several control experiments, the results of which are described in Sieprath et al.1. In brief, the fluorescence response of CM-H2DCFDA and TMRM to extraneously induced changes in intracellular ROS and Δψm, respectively has been quantified to determine the dynamic range. For CM-H2DCFDA, NHDF showed a linear increase in fluorescence signal when treated with increasing concentrations of TBHP between a range of 10 µM to 160 µM. Likewise, for TMRM, NHDF cells demonstrated a linear increase in mitochondrial fluorescence when treated with increasing concentrations of oligomycin (which induces Δψm hyperpolarization) within a 1 to 10 µg/µL range. Conversely, real-time addition of valinomycin, an antibiotic that induces Δψm depolarization, resulted in a gradual, quantifiable decrease of TMRM fluorescence.
The assay has also been used in a variety of experiments to reveal differences in redox status between cell types or treatments1,15. To illustrate this, the results are shown of an experiment in which NHDF were treated with the HIV protease inhibitor Saquinavir (SQV) (Figure 5). Using the described protocol, a significant increase was detected for both basal and induced ROS levels as compared to control cells treated with DMSO (Figure 5B). SQV treatment also significantly affected mitochondrial morphofunction. Morphologically, mitochondria acquired a highly fragmented pattern, which was also confirmed by a higher circularity and smaller average size of the individual mitochondria. Functionally, Δψm, measured as average TMRM signal per mitochondrial pixel was also significantly increased (Figure 5A). When combining the data of the 5 parameters described above, the two conditions (control and SQV) could be clearly separated from each other by principal component analysis. Data from three independent biological replicates is shown in a 2D-biplot displaying the first two principal components, which explain 81.4% of the total variance (Figure 5C). This proves the robustness of the assay and suggests that the combined readout may serve as a sensitive indicator of cellular health status.
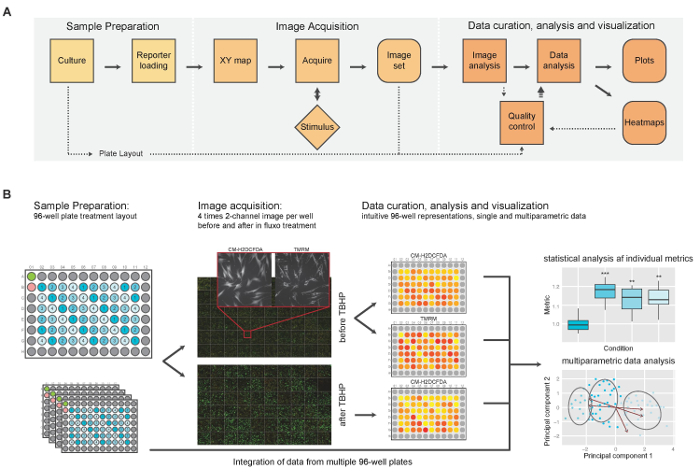
Figure 1: General Overview of the High-content Imaging Assay for Simultaneous Measurement of Intracellular Basal and Induced ROS Levels and Mitochondrial Morphofunction. (A) Schematic representation of the major operational blocks. (B) Illustrated example: cells are seeded in multiple identical 96-well plates. A standard well plate layout is shown in more detail in Figure 2A. After staining, 4 images are acquired per channel around the center of each well, both pre and post TBHP treatment, which is illustrated by the large montages with inset. After image analysis, intensity results are visualized using an intuitive heat-map projected onto the well-plate layout. This permits rapid detection of plate effects or aberrant wells. After curation of the complete experimental data sets, final data analysis is performed resulting in single- as well as multi-parameter output. (This figure was modified from reference1, with permission of Springer) Please click here to view a larger version of this figure.
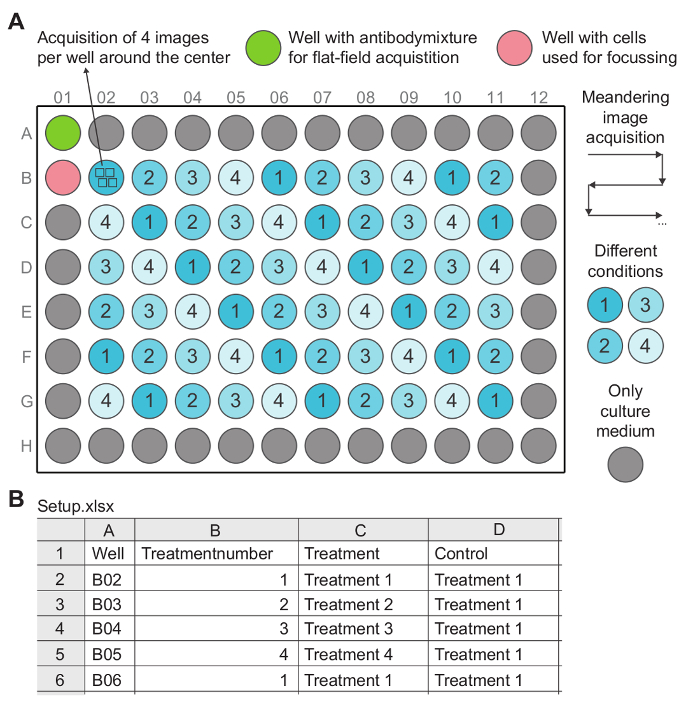
Figure 2: A Typical Experimental 96-well Layout and Corresponding 'Setup' File. (A) 4 different conditions are distributed homogeneously across the inner 60 wells of the plate. Well B01 also contains cells, but is only used to adjust the initial PFS offset just before imaging. The other outer wells are filled only with culture medium to minimize gradients (temperature, humidity, etc.) during cell culture. Image acquisition is performed in a meandering manner, i.e., first from left to right, from well B02 to B11, then back, from right to left, from well C11 to C02 and so on. After image acquisition flat field images are acquired in well A01, which is used for correcting spatial illumination heterogeneity during image analysis. (B) The corresponding setup file (Setup.xlsx) is a spreadsheet that contains information about the layout of the experiment. It specifies the locations of each treatment in the multiwell plate and their respective controls. Each row represents a well. Each treatment has its own unique treatment number, which is used to specify the order of the treatments on the X-axis of the generated plots during data analysis. The 'Control' column contains the treatment that should be used as a control for normalizing the data of the treatment specified in the same row. In the example, treatment 1 is the treatment that is used as control for normalizing the data from all the other treatments. Please click here to view a larger version of this figure.
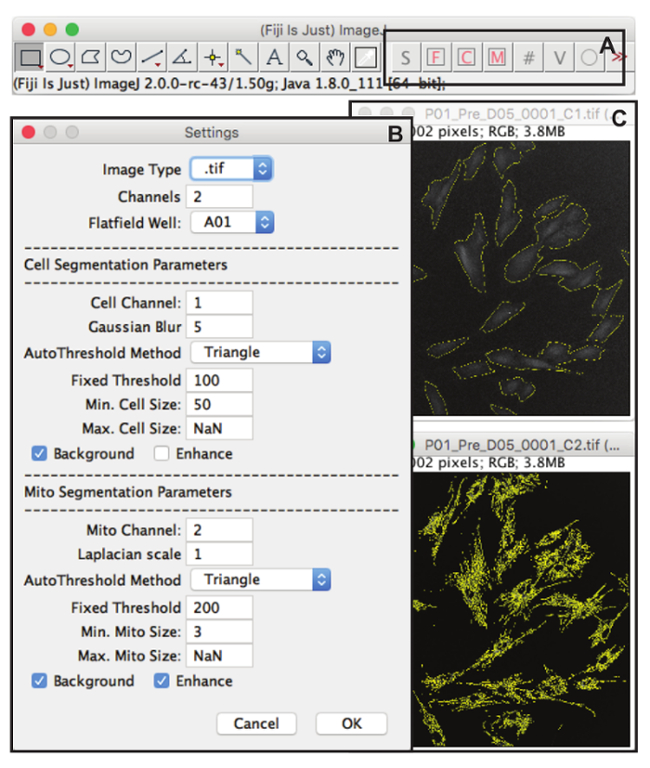
Figure 3: RedoxMetrics Macro-set, Layout and Setup Interface. (A) When the RedoxMetrics macro-set for FIJI is installed, a number of new macro commands as well as a set of action tools to test and optimize the analysis settings is created in the menu bar. 'S' invokes the setup interface. 'F' performs a flatfield correction on an open image. 'C' and 'M' perform a test segmentation for cellular regions ("Cell", CM-H2DCFDA channel) and mitochondrial regions ("Mito", TMRM channels), respectively on a single opened image using the analysis settings selected in the setup interface, returning an overlay of the segmented regions of interest (ROIs). '#' performs batch analysis on a folder of images using the settings specified in the setup interface (usually after verification on one or a few images). 'V' creates a verification hyperstack using the output data from the batch analysis. This hyperstack is a composite image of all raw images present in the folder and their respective regions of interest (drawn in a second channel). 'O' can be clicked to show or hide the overlay of the segmented region. (B) Setup interface. Here all analysis settings are selected, including general information such as the image type (extension), number of channels (wavelengths), and the well that was used for flatfield correction. Next there are settings specific to the image content type (Cell or Mito). In both cases, there are options (checkboxes) to include a background correction ("background") and local contrast enhancement ("contrast"). For cell segmentation, there is the option to define a Gaussian blur radius (sigma) for reducing noise. For mito segmentation there is the option to define the radius of a Laplacian operator for selective enhancement of the mitochondria. Subsequent segmentation is performed using an automatic (or fixed) thresholding method that can be defined per content type. When a fixed threshold is chosen, the upper threshold value can be provided manually. Finally, the analysis can be restricted to a selection of objects that falls within a minimum and max size. (C) An example of a segmentation result as run with the "C" (top) or "M" (bottom) command, displayed as the raw image in grey overlaid with the regions of interest in yellow. Please click here to view a larger version of this figure.
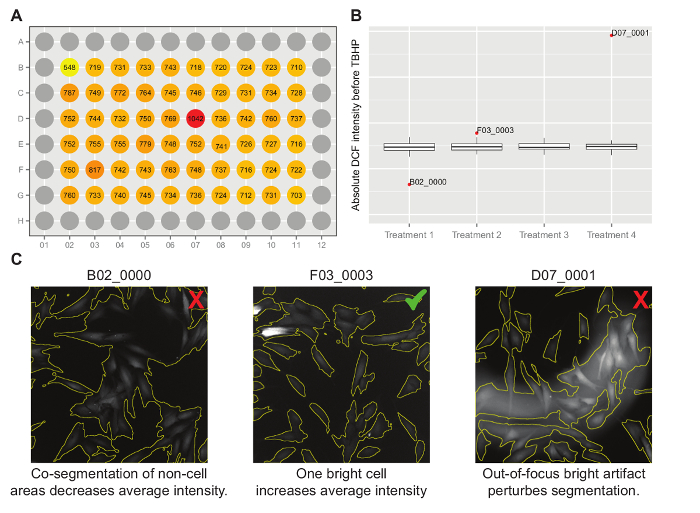
Figure 4: Illustrated Example of Intermediate Data Visualizations. (A) Multiwell plate visualization where wells B02 and D07 appear suspicious. (B) Boxplot representing the same data, with the outliers labeled with well name and image name. Together with F03_0003, images from B02 and D07 reappear as outliers. (C) Visual inspection of these images shows that B02_0000 and D07_0001 should be removed from further analysis because there are segmentation errors (illustrated by the red 'X'). F03_0003 should be kept because there is no apparent segmentation or technical error. (if no technical reason is found for the aberration, the data should not be removed). Please click here to view a larger version of this figure.
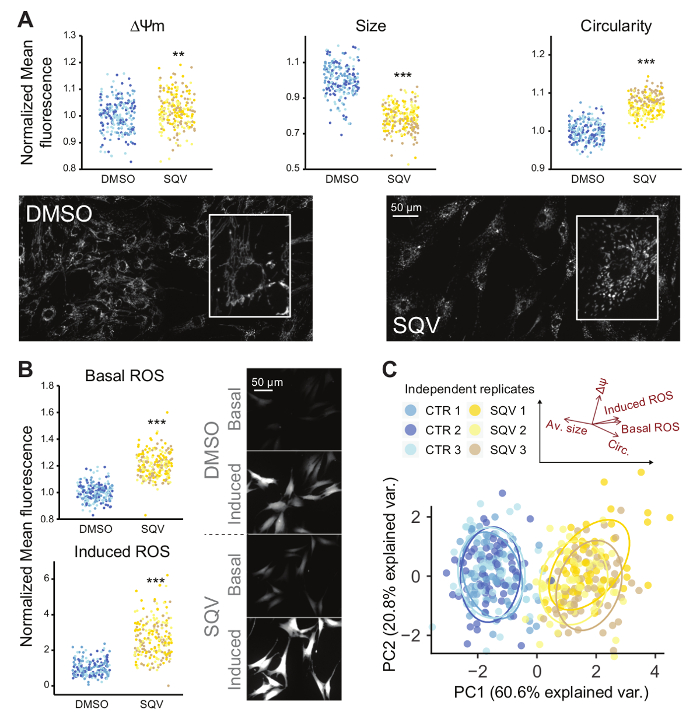
Figure 5: Effect of Saquinavir (SQV; 20 µM) on Primary Human Fibroblasts (control is DMSO). (A) Mitochondrial membrane potential (ΔΨm) and mitochondrial morphology (circularity and average size of individual mitochondria) as measured by TMRM (B) Increased Basal levels of intracellular ROS as measured by CM-H2DCFDA and response towards induced ROS, measured as relative gain in intensity after addition of 20µM TBHP addition. (C) 2D scatterplot of the first 2 principal components (PCs) from a PCA analysis on the 5 variables described above (basal and induced ROS levels, average mitochondrial size, circularity, and ΔΨm). The black arrows represent the directions of the original 5 variables with respect to the principal components. (Independent replicates are plotted with a different color; All data is normalized with respect to the DMSO-control; * = p value <0.05; ** = p value <0.01; *** = p value <0.001; the range of the Y -axes has been adjusted to optimally display the differences; This figure was modified from reference1, with permission of Springer) Please click here to view a larger version of this figure.
| Problem | Potential reason | Possible solution |
| Too few cells in the wells during imaging | Too few cells seeded | Seed more cells 24 h before imaging |
| Poor cell growth | Check culture conditions (medium, temperature stability, CO2 control) | |
| Too violent washing steps | Avoid washing cells away, pipette gently | |
| Too many cells in the wells during imaging | Too many cells seeded | Seed less cells 24 h before imaging |
| Weak signals or non-linear response of CM-H2DCFDA fluorescence to stimulus | Used too low/high dye concentration | If using other than NHDF cells, optimal loading concentrations have to be determined empirically |
| Dye-loading is insufficient | Add 0.02% w/v of the surfactant Pluronic-127 to the loading solution to increase uptake. | |
| CM-H2DCFDA signal visually increases during exposure time | Too high dye concentration | Reduce dye concentration |
| Excitation intensity is too high | Reduce excitation intensity by using ND filters and/or reducing exposure time | |
| CM-H2DCFDA intensity decreases during acquisition of the well plate | CM-H2DCFDA is leaking out of the cell. | Use 2 mM Probenicid (anion pump inhibitor). Add to the loading solution, as well as the imaging buffer to decrease leakage |
| There is no increase in CM-H2DCFDA signal intensity after addition of TBHP | TBHP is not active | Use fresh TBHP |
| CM-H2DCFDA is leaking out of the cell. | Use Probenicid as described above. | |
| (Some of the) acquired images appear out-of-focus, while focus seems ok when checked. | The plate is mounted tilted causing the focus to extend beyond the PFS offset | Check the mounting of the plate and the stage level, Reposition the plate |
| The bottom of the plate contains dust or irregularities | Clean the bottom of the plate with an ethanol-wetted lens paper | |
| Cells die during imaging | Excitation intensity is too high | Reduce excitation intensity or acquire fewer images per well. |
| Wrong imaging-buffer composition | Remake imaging-buffer | |
| There are out of focus fluorescent spots in the images | Detached cells are floating out of focus | Wash more gently during the loading protocol |
| One or more columns have an allover lower intensity than the other wells of the plate | One or more tips from the multichannel pipette were not firmly attached and therefore less reporter was added to these wells | Thoroughly check if all tips are filled perfectly whenever you use a multichannel pipette. |
| Focus drifts dramatically during image acquisition | The polystyrene bottom of the plate can react with immersion oil when using an oil lens | Use a dry lens, or use a multiwell plate with a glass bottom |
| TMRM signal intensity increases between imaging the first and last well | Reporter loading time was not long enough, TMRM is still equilibrating | Increase reporter loading time |
| CM-H2DCFDA signal after TBHP treatment increases during acquisition of the well plate | The time between treatment with TBHP and the start of the second imaging round was not long enough, TBHP is still oxidizing CM-H2DCFDA. | Increase incubation time between the treatment with TBHP and the second imaging round. (start with at least 3 min) |
| Flat field images are saturated | Antibody working solution is to concentrated | Use a lower concentration of antibody working solution |
| Acquisition settings are not optimized (exposure time to high, ND-filter to low, …) | Optimize acquisition settings, signal has to be in dynamic range of the sensor | |
| Flat field images are to dark | Antibody working solution is to diluted | Use a higher concentration of antibody working solution |
| Acquisition settings are not optimized (exposure time to high, ND-filter to low, …) | Optimize acquisition settings, signal has to be in dynamic range of the sensor | |
| Over/Undersegmentation artefacts | Threshold not set correctly | Adjust the threshold setting |
| Size filters not adjusted properly | Adjust the min and max size criteria for image analysis | |
| Cell confluency is too high | The correct level of confluency is very important for reliable image analysis. For other cell types than NHDF, the optimal seeding density has to be determined empirically | |
| The shiny application does not work | Wrong directory selected | Choose the correct directory on the input page. |
| Missing files in directory | Make sure all necessary files (output from imagej macro and setup file) are present in the selected directory | |
| Missing packages | Normally the app checks for and installs all necessary packages, but when this fails, check manually for the following packages: devtools, ggbiplot, pacman, ggplot2, plyr, nparcomp, data.table, readxl, gplots, ggbiplot, shiny, shinyFiles, pbapply and shinyjs including all dependencies. |
Table 1: Typical pitfalls and potential solutions.
